Predicting First-Pass Metabolism with PBPK Modeling: A Guide for Drug Development Researchers
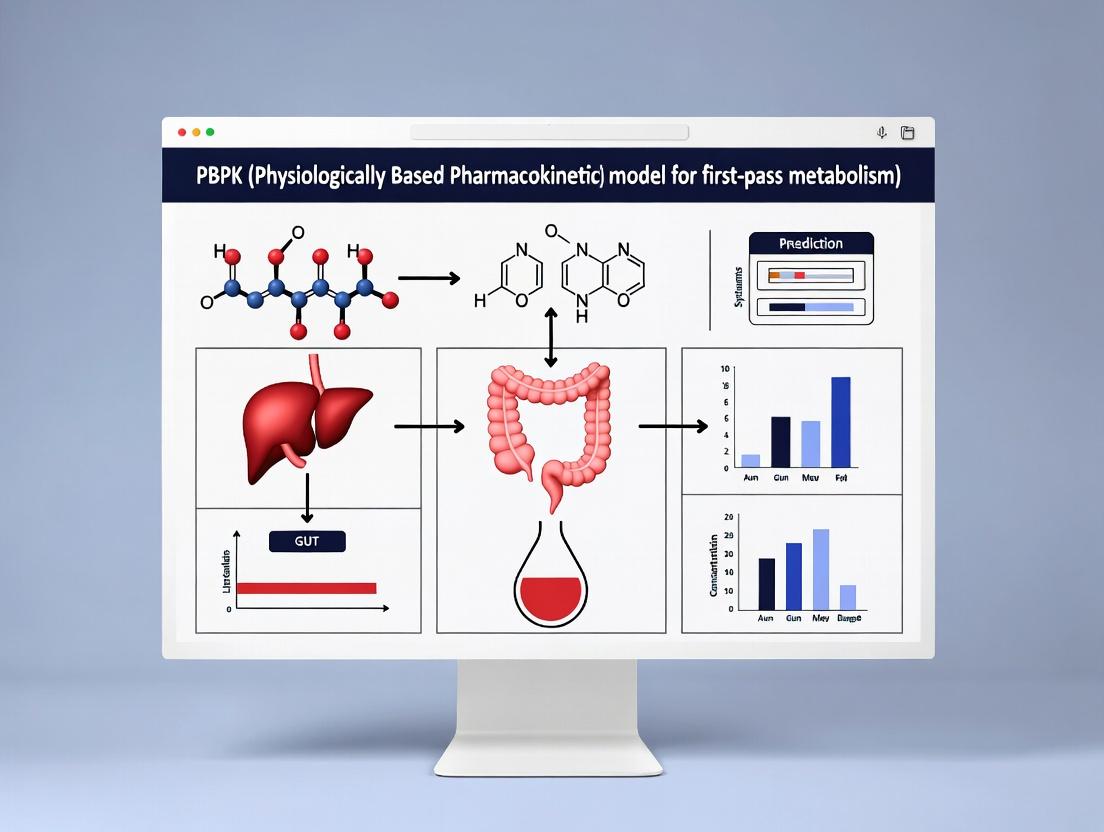
This article provides a comprehensive guide for researchers and drug development professionals on leveraging Physiologically Based Pharmacokinetic (PBPK) modeling to predict first-pass metabolism.
Predicting First-Pass Metabolism with PBPK Modeling: A Guide for Drug Development Researchers
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on leveraging Physiologically Based Pharmacokinetic (PBPK) modeling to predict first-pass metabolism. It explores the foundational concepts of hepatic and intestinal extraction, details the methodological steps for model construction and application, addresses common troubleshooting and optimization challenges, and reviews validation standards and comparative analyses with traditional methods. The content synthesizes current best practices to enhance the predictive accuracy of oral bioavailability and streamline drug candidate selection.
Understanding First-Pass Metabolism: The Critical Role of PBPK Modeling Fundamentals
This technical support center provides troubleshooting guidance and FAQs for researchers conducting in vitro and in vivo experiments to quantify first-pass metabolism for PBPK (Physiologically Based Pharmacokinetic) model development. The content is framed within the thesis: "Advancing the Predictivity of PBPK Models for First-Pass Metabolism Through Integrated In Vitro-In Vivo Extrapolation (IVIVE)."
FAQs & Troubleshooting
Q1: Our PBPK model consistently underestimates the oral AUC of our test compound. What are the primary experimental sources of this error? A: This often stems from an incomplete accounting of extraction sites. Key troubleshooting steps:
- Check Intestinal Assays: Confirm your model includes both gut wall metabolism (e.g., using human intestinal microsomes or expressed CYP3A4) and consideration of gut luminal degradation. Neglecting intestinal extraction is a common oversight.
- Verify Hepatic Parameters: Re-examine the determination of hepatic intrinsic clearance (CLint). Ensure in vitro incubations (using human liver microsomes or hepatocytes) used appropriate protein binding corrections (fu,inc) and accounted for non-microsomal enzymes if relevant.
- Assess Assumption of Sequential Extraction: The classic "gut-liver" sequential model may not hold for all compounds. Consider experimental designs to dissect simultaneous extraction.
Q2: When using human hepatocytes in suspension to measure hepatic CLint, we observe high inter-donor variability. How do we determine a representative value for PBPK input? A: This is expected due to genetic polymorphisms. The recommended protocol is:
- Use a Minimum of 10 Donor Pools: Source hepatocytes from a pool of 10 or more individual donors to capture population variability.
- Run Concentration-Dependence: Perform substrate depletion or metabolite formation assays across a range of clinically relevant concentrations.
- Data Analysis: Calculate CLint for each donor pool. Use the geometric mean of the individual CLint values for the "average" population input. To model variability, incorporate the observed standard deviation into a population PBPK simulation.
Q3: What is the most robust experimental workflow to deconvolve the relative contributions of intestinal vs. hepatic first-pass extraction for a new chemical entity? A: An integrated in vivo pharmacokinetic study in preclinical species (e.g., rat) with surgical modifications, followed by in vitro IVIVE.
- Protocol: Compare systemic exposure (AUC) after four administration routes in a crossover design: intravenous (IV), intraportal (IPV), intra-arterial (IA), and oral (PO).
- Calculations: Use the following equations to derive extraction ratios:
| Route Comparison | Calculation | Extraction Site Quantified |
|---|---|---|
| EH (Hepatic) | 1 – (AUCIPV / AUCIA) | Liver |
| EG (Gut) | 1 – (AUCPO / AUCIPV) | Intestinal Wall |
| FH | 1 – EH | Hepatic Availability |
| FG | 1 – EG | Gut Wall Availability |
| Overall F | FG x FH | Total Oral Bioavailability |
Q4: How do we translate in vitro Michaelis-Menten parameters (Vmax, Km) from recombinant enzyme systems into organ-specific extraction ratios for a PBPK model? A: The critical step is scaling via the RAF/ISEF approach (Relative Activity Factor/Inter-System Extrapolation Factor).
- Protocol: Conduct parallel incubation experiments using (a) the recombinant enzyme (rCYP) and (b) human liver microsomes (HLM) with a selective probe substrate for that enzyme (e.g., midazolam for CYP3A4).
- Calculation:
- Determine the Vmax for the probe in both systems.
- Calculate RAF = (Vmax (probe in HLM)) / (Vmax (probe in rCYP)).
- Apply this RAF to scale the Vmax of your test compound from the rCYP system to the microsomal system: Vmax,HLM = Vmax,rCYP x RAF.
- Use the scaled Vmax,HLM and the measured Km to calculate CLint, which is then scaled to whole liver using physiological scaling factors (microsomal protein per gram of liver, liver weight).
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in First-Pass Metabolism Research |
|---|---|
| Cryopreserved Human Hepatocytes (Suspended & Plated) | Gold-standard system for measuring integrated phase I/II hepatic metabolism and active uptake/efflux; used for CLint determination and transporter studies. |
| Human Liver Microsomes (HLM) & Intestinal Microsomes (HIM) | Pooled from multiple donors; contain cytochrome P450 and UGT enzymes for efficient, high-throughput measurement of metabolic stability and reaction phenotyping. |
| Recombinant CYP/UGT Enzymes (rCYP) | Expressed singly in insect or mammalian cells; essential for reaction phenotyping to identify the specific enzyme(s) responsible for metabolism and for RAF/ISEF scaling. |
| Transfected Cell Systems (e.g., MDCK, HEK293 expressing OATP1B1, P-gp, BCRP) | Used in bidirectional transport assays to quantify the role of specific uptake and efflux transporters in hepatic and intestinal extraction. |
| Specific Chemical & Antibody Inhibitors | Used in in vitro incubations to phenotypically assess the contribution of specific enzymes (e.g., ketoconazole for CYP3A4) or transporters (e.g., cyclosporine A for OATPs/P-gp). |
| Semi-permeable Membrane Devices (e.g., Caco-2 cells, PAMPA) | Models of intestinal permeability to predict fraction absorbed (Fa) and assess transporter effects in the gut. |
Experimental Protocols & Data Presentation
Protocol 1: Determination of Hepatic Intrinsic Clearance (CLint) using Human Hepatocytes in Suspension.
- Thawing: Rapidly thaw cryopreserved hepatocytes in a 37°C water bath and transfer to pre-warmed recovery medium.
- Incubation: After cell viability assessment (>80%), suspend hepatocytes (0.5 x 10⁶ cells/mL) in incubation buffer. Pre-incubate at 37°C for 5 min.
- Reaction: Initiate reaction by adding test compound (at least 5 concentrations spanning expected Km). Perform incubations in triplicate.
- Termination: At predetermined time points (e.g., 0, 5, 15, 30, 45, 60 min), remove aliquots and quench with acetonitrile containing internal standard.
- Analysis: Centrifuge, analyze supernatant via LC-MS/MS to determine parent compound depletion.
- Data Processing: Fit depletion curves to a first-order decay model: CLint, vitro = k (depletion rate constant) / (Number of cells per mL x Cell volume). Scale to whole liver using 120 x 10⁶ cells per gram liver and 21 g liver weight per kg body weight.
Table 1: Example In Vitro to In Vivo Scaling of Hepatic Clearance
| Parameter | Value | Source/Calculation |
|---|---|---|
| In vitro CLint (µL/min/million cells) | 25.4 | Measured in hepatocyte depletion assay |
| Scaling Factor (million cells/g liver) | 120 | Physiological scalar |
| Liver Weight per kg BW (g/kg) | 21 | Physiological scalar |
| Predicted Hepatic CLint (mL/min/kg) | 64.0 | = 25.4 * 120 * 21 / 1000 |
| Predicted Hepatic Blood Flow (mL/min/kg) | 21 | Species-specific (human) |
| Predicted Hepatic Extraction Ratio (EH) | 0.75 | = 64 / (64 + 21) [Well-Stirred Model] |
Protocol 2: Assessing Gut Wall Metabolism using Human Intestinal Microsomes (HIM).
- Incubation Setup: Prepare HIM (0.1-0.5 mg protein/mL) in potassium phosphate buffer with MgCl₂.
- Pre-incubation: Add NADPH-regenerating system and pre-incubate at 37°C for 5 min.
- Reaction Initiation: Add substrate (at pharmacologically relevant concentration). Run in triplicate with negative controls (no NADPH).
- Termination & Analysis: Quench at time points (e.g., 0, 10, 20, 30 min) with cold acetonitrile. Analyze via LC-MS/MS for metabolite formation or substrate depletion.
- Data Processing: Calculate CLint, micro (µL/min/mg protein) from initial linear rates. Scale using intestinal scaling factors: Intestinal CLint = CLint, micro x MPPGLI x Intestinal Weight, where MPPGLI is microsomal protein per gram of intestine (~15 mg/g).
Visualizations
Title: First-Pass Extraction: Sequential Gut-Liver Model
Title: Integrated IVIVE Workflow for PBPK Model Development
This technical support center is framed within a thesis investigating PBPK modeling to predict first-pass metabolism. It provides targeted troubleshooting and FAQs for researchers, scientists, and drug development professionals implementing these critical models.
Troubleshooting Guides & FAQs
Q1: My model consistently under-predicts oral bioavailability for a high-permeability drug. Which parameters should I investigate first? A: This often points to an inaccurate estimation of first-pass intestinal or hepatic extraction.
- Check Intestinal Metabolism: Verify the enterocyte concentration of relevant CYP enzymes (e.g., CYP3A4) and the scaling factor used.
- Validate Hepatic Clearance: Ensure the intrinsic clearance (CLint) value, derived from in vitro hepatocyte assays, is appropriately scaled using accurate liver weight and microsomal or hepatocyte protein yield.
- Review Blood Flow Rates: Confirm the hepatic portal vein and arterial blood flow rates in your physiological model are appropriate for your species and subject demographics.
- Protocol: In vitro-in vivo extrapolation (IVIVE) for Hepatic CLint
- Objective: To scale in vitro metabolic stability data to an in vivo hepatic clearance value.
- Method: Incubate the drug with pooled human liver microsomes (HLM) or hepatocytes at relevant concentrations. Determine the in vitro half-life and calculate in vitro CLint.
- Calculation: Apply the "well-stirred" liver model: CLh = (Qh * fu * CLintin vitro) / (Qh + fu * CLintin vitro), where Qh is hepatic blood flow, and fu is the fraction unbound in blood.
Q2: During model validation, systemic clearance is accurate, but the predicted plasma concentration-time profile shape is wrong. What could be the issue? A: The mismatch in profile shape with accurate AUC suggests a mis-specification of distributional parameters.
- Troubleshoot Tissue Partitioning: The most common cause. Review the method used to calculate tissue-to-plasma partition coefficients (Kp). The Poulin and Rodgers (lipid composition) method may fail for ionized or specialized transporter substrates.
- Check Absorption Kinetics: For oral dosing, an incorrect absorption rate constant (Ka) can distort the early profile. Consider using a double-peak function if enterohepatic recirculation is suspected.
Q3: How do I model a prodrug where hydrolysis occurs in the gut lumen prior to absorption of the active moiety? A: This requires a multi-compartment absorption model.
- Structure: Model the prodrug as a separate species in the gut lumen compartment.
- Process: Include a first-order or enzymatic conversion rate from prodrug to active drug within the gut lumen compartment.
- Absorption: Link the converted active drug to the standard absorption pathway into the enterocyte. The conversion rate constant must be estimated from in vitro simulated intestinal fluid stability studies.
Key Compartments & Parameters: Data Tables
Table 1: Essential Physiological Compartments in a Full PBPK Model
| Compartment | Description | Key Physiological Parameters (Human, 70kg) | Relevance to First-Pass Metabolism |
|---|---|---|---|
| Lung | Often included as a mixing chamber. | Blood flow: ~100% of Cardiac Output | Low affinity binding can affect initial distribution. |
| Liver | Major site of metabolism and biliary excretion. | Blood flow: ~1.55 L/min; Weight: ~1.5 kg; CYP enzyme abundances (pmol/mg protein). | Primary organ for systemic and first-pass hepatic clearance. |
| Gut (Lumen & Enterocytes) | Site of absorption and intestinal metabolism. | pH gradient (stomach 1.5-3, intestine ~6.5); Transit times; CYP3A4 abundance in enterocytes. | Governs fraction absorbed and pre-systemic intestinal extraction. |
| Kidney | Organ of renal excretion. | Blood flow: ~1.2 L/min; Glomerular Filtration Rate (GFR): ~120 mL/min. | Accounts for renal clearance. |
| Adipose & Muscle | Large distribution volumes. | Tissue volumes, perfusion rates, composition (neutral lipid, water content). | Determine the volume of distribution and terminal phase. |
| Venous & Arterial Blood | Central blood pools for mass balance. | Plasma volume, blood-to-plasma ratio, hematocrit. | Driving force for perfusion-limited distribution. |
Table 2: Critical Drug-Dependent Parameters for Absorption & Clearance
| Parameter | Symbol | Typical Source/Assay | Impact on Model Prediction |
|---|---|---|---|
| Effective Permeability | Peff | Caco-2 assay, MDCK cells, or in situ perfusion. | Directly determines absorption rate constant (Ka) and fraction absorbed. |
| Solubility (pH-dependent) | S | Shake-flask or biorelevant dissolution (FaSSIF/FeSSIF). | Limits maximum dissolved dose, critical for Biopharmaceutics Classification System (BCS) II/IV drugs. |
| Intrinsic Clearance | CLintin vitro | Hepatocyte or microsomal stability incubation. | Scaled to in vivo hepatic metabolic clearance. |
| Fraction Unbound in Blood/Plasma | fu / fu,p | Equilibrium dialysis or ultracentrifugation. | Determines free drug available for metabolism/distribution. |
| Tissue-to-Plasma Partition Coefficients | Kp | In silico prediction (e.g., Rodgers & Rowland), in vivo tissue sampling. | Dictates the extent of drug distribution into tissues. |
| Biliary Clearance | CLbile | Sandwich-cultured hepatocyte assay. | Predicts fecal excretion and potential enterohepatic recirculation. |
Experimental Protocols for Key Inputs
Protocol 1: Determining Effective Permeability (Peff) using Caco-2 Monolayers
- Objective: To estimate human intestinal permeability in vitro.
- Materials: Caco-2 cells (passage 40-60), Transwell inserts, transport buffers (pH 7.4, 6.5), LC-MS/MS for quantification.
- Method:
- Seed cells on inserts and culture for 21 days to form confluent, differentiated monolayers. Confirm integrity via TEER (>300 Ω·cm²).
- Add drug to donor compartment (apical for A→B, basolateral for B→A). Use a low-solubility control (e.g., atenolol) and high-permeability control (e.g., propranolol).
- Sample from the receiver compartment at intervals over 2 hours.
- Calculate apparent permeability: Papp = (dQ/dt) / (A * C0), where dQ/dt is the transport rate, A is the filter area, and C0 is the initial donor concentration.
- Apply a correlation (e.g., from literature) to scale Papp to human in vivo Peff.
Protocol 2: Measuring Fraction Unbound (fu) via Equilibrium Dialysis
- Objective: To determine the unbound fraction of drug in plasma.
- Materials: Equilibrium dialysis device, dialysis membranes (MWCO 12-14 kDa), blank plasma, phosphate buffer (pH 7.4).
- Method:
- Spike drug into plasma side to a therapeutically relevant concentration.
- Assemble the device with buffer on the other side of the membrane.
- Dialyze at 37°C with gentle rotation for 4-6 hours (validate time to equilibrium).
- Post-dialysis, quantify drug concentration in both plasma and buffer chambers using a sensitive assay (LC-MS/MS).
- Calculate fu = [Drug]buffer / [Drug]plasma. Apply a volume shift correction if necessary.
Visualizing the PBPK Structure and First-Pass Pathways
PBPK Model Structure and First Pass Pathway
PBPK Input to Output Workflow for First Pass
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in PBPK-Related Research |
|---|---|
| Pooled Human Liver Microsomes (HLM) | Source of CYP enzymes for in vitro intrinsic clearance (CLint) determination and metabolite identification. |
| Cryopreserved Human Hepatocytes | Gold-standard cell system for predicting hepatic CLint, transporter effects, and biliary clearance. |
| Caco-2 Cell Line | Model for predicting human intestinal permeability and investigating active transport/efflux. |
| Simulated Intestinal Fluids (FaSSIF/FeSSIF) | Biorelevant media for assessing dissolution and solubility under physiological conditions. |
| Equilibrium Dialysis Plates | High-throughput method for determining plasma protein binding (fu). |
| LC-MS/MS System | Essential for sensitive and specific quantification of drugs and metabolites in in vitro and in vivo samples. |
| PBPK Software (e.g., GastroPlus, Simcyp, PK-Sim) | Platform for integrating in vitro and physiological data to build, simulate, and validate models. |
| CYP-Specific Chemical Inhibitors (e.g., Ketoconazole) | Used in in vitro reaction phenotyping to identify enzymes responsible for metabolism. |
Technical Support Center: Troubleshooting PBPK-Focused Intestinal Metabolism Experiments
FAQs & Troubleshooting Guides
Q1: In our intestinal S9 fraction incubations, metabolite formation is lower than predicted, and variability is high. What are the primary causes and solutions? A: This commonly stems from improper handling of subcellular fractions or cofactor depletion.
- Troubleshooting Steps:
- Verify Fraction Integrity: Confirm protein concentration via Bradford assay and check for signs of degradation (e.g., repeated freeze-thaw >2 cycles). Use fresh or single-thaw aliquots.
- Optimize Cofactor Regeneration System: For Phase I (CYP450), ensure NADPH is fresh and the regeneration system (e.g., Glucose-6-Phosphate, G6PDH) is active. For Phase II (UGTs), ensure UDPGA is fresh and in sufficient molar excess.
- Incorporate Transporter Inhibition: If using intact systems (like suspended enterocytes), consider adding a broad transporter inhibitor (e.g., 100 µM verapamil) to distinguish metabolism from efflux-limited access.
- Protocol: Intestinal S9 Fraction Incubation for Intrinsic Clearance (CLint)
- Reagents: Pooled human intestinal S9 fraction (commercial source), 1 mM NADPH regenerating system (1.3 mM NADP+, 3.3 mM G6P, 0.4 U/mL G6PDH, 3.3 mM MgCl₂), 100 mM phosphate buffer (pH 7.4), test compound (substrate).
- Method:
- Pre-incubate S9 (0.2-0.5 mg protein/mL) with substrate (1 µM) in buffer at 37°C for 5 min.
- Initiate reaction by adding full NADPH regenerating system.
- Aliquot at 5-6 time points (e.g., 0, 5, 10, 20, 30, 45 min). Terminate with 2 vols of ice-cold acetonitrile containing internal standard.
- Centrifuge (3000g, 15 min, 4°C). Analyze supernatant via LC-MS/MS for parent depletion/metabolite formation.
- Calculate in vitro CLint from the slope of the natural log of substrate depletion over time.
Q2: When using Caco-2 or induced pluripotent stem cell-derived enterocyte models for permeability and metabolism studies, how do we deconvolute the contribution of efflux transporters (like P-gp) from CYP3A4 metabolism? A: This requires a strategic combination of chemical inhibitors and experimental design.
- Troubleshooting Steps:
- Bidirectional Transport with Inhibitors: Conduct standard bidirectional (A-to-B, B-to-A) assays with and without specific inhibitors.
- Sequential Inhibition: First, use a potent P-gp inhibitor (e.g., 10 µM zosuquidar) to isolate the permeability component. Then, add a CYP3A4 inhibitor (e.g., 1 µM ketoconazole) to assess the metabolic component in the absence of efflux.
- Measure Metabolites: Quantify major metabolites in both donor and receiver chambers to track metabolic fate alongside transport.
- Key Data Table: Inhibitor Concentrations for Deconvolution Studies
Target Example Inhibitor Recommended Concentration (in vitro) Primary Use P-glycoprotein (P-gp) Zosuquidar (LY335979) 5-10 µM Inhibit drug efflux transport CYP3A4 Ketoconazole 1 µM Inhibit oxidative metabolism BCRP Ko143 1-5 µM Inhibit efflux transport All Major CYP450s 1-Aminobenzotriazole (ABT) 1 mM (pre-incubation) Mechanism-based inactivation
Q3: We are developing a PBPK model for first-pass metabolism. What are the critical in vitro to in vivo extrapolation (IVIVE) scaling factors for intestinal CYP3A4, and why might scaled clearance still under-predict in vivo extraction? A: Under-prediction often arises from overlooking enterocyte biology and sequential processes.
- Troubleshooting & Key Considerations:
- Scaling Factor Consistency: Ensure you use the correct scaling factors consistently.
- S9 Scaling: ISEF (Intersystem Extrapolation Factor) for specific CYP isoforms and SFu (Fraction unbound in incubation) are critical.
- Whole-cell Scaling: Use cellularity (enterocytes per gram intestine) and total intestinal mucosal mass.
- Sequential Metabolism & Transporter Interplay: Intracellular metabolism can be limited by uptake (e.g., via OATP2B1) or enhanced by efflux (P-gp) causing metabolite re-entry into CYP3A4. Your PBPK model must account for this interplay.
- Villi Blood Flow & Permeability: The effective permeability (Peff) and villous blood flow rate are key determinants of substrate access to enterocytes. Validate your Peff values.
- Scaling Factor Consistency: Ensure you use the correct scaling factors consistently.
- Table: Key Scaling Factors for Intestinal IVIVE in PBPK
Parameter Symbol Typical Value (Human) Source/Note Intestinal Tissue Density ρ 1.05 g/mL Assumed Microsomal Protein per g Intestine MPPGI 30-40 mg/g Lot-to-lot variability; use lot-specific S9 Protein per g Intestine S9PPGI ~65 mg/g From histology & protein content Enterocyte Cellularity #Cells/g 100-130 million cells/g Derived from villus geometry Intestinal Mucosal Mass ~200 g (adult) Age- and population-dependent Fraction Unbound in Incubation SFu Determined experimentally Use measured fu_inc
The Scientist's Toolkit: Key Research Reagent Solutions
| Item / Reagent | Function in CYP450/Transporter Studies |
|---|---|
| Pooled Human Intestinal Microsomes/S9 | Contains native complement of CYP450s and UGTs for intrinsic clearance assays. |
| Transfected Cell Systems (e.g., MDCK-OATP2B1+P-gp) | Engineered to express single transporters for mechanistic uptake/efflux studies. |
| Induced Pluripotent Stem Cell (iPSC)-Derived Enterocytes | Physiologically relevant model with co-expressed metabolizing enzymes and transporters. |
| LC-MS/MS with High Sensitivity | Essential for quantifying low-abundance metabolites and parent drug in complex matrices. |
| Stable Isotope-Labeled Substrates (e.g., ¹³C-Verapamil) | Used as internal standards or probes to track specific metabolic pathways without interference. |
| Selective Chemical Inhibitors (see Table above) | To deconvolute the contribution of specific enzymes or transporters in complex systems. |
| NADPH Regeneration System (lyophilized) | Ensures consistent cofactor supply for oxidative metabolism during longer incubations. |
Visualizations
Title: IVIVE Workflow for Intestinal Metabolism in PBPK
Title: Drug Fate in Enterocyte: Enzymes & Transporters
Troubleshooting Guides & FAQs
Q1: Our PBPK model consistently underpredicts oral bioavailability for compounds known to be high CYP3A4 substrates. What could be the cause? A: This is often due to inaccurate characterization of intestinal first-pass metabolism. Key troubleshooting steps include:
- Verify enzyme abundance data: Ensure you are using the most recent, tissue-specific abundance values for CYP3A4 in the gut wall (e.g., from Paired Intestine-Liver samples). Older models may use outdated scalars.
- Check enterocyte transit time: The default intestinal transit time may be too rapid. Consider implementing a more refined model that accounts for regional differences in permeability and metabolism.
- Confirm inhibition constants (Ki): Re-evaluate in vitro Ki values for any potential self-inhibition or food-component interactions that may not be adequately modeled.
- Validate hepatic influx: For some compounds, hepatic uptake (via OATP transporters) can be rate-limiting and if mis-specified, can skew the apparent first-pass contribution.
Q2: How do we handle variability in gut microbiome metabolism when predicting bioavailability? A: Microbiome metabolism is an emerging source of variability. Current best practices are:
- Incorporate as a discrete variable: Model it as a binary (high/low metabolizer) or categorical variable in sensitivity analysis or virtual population simulations.
- Use biorelevant in vitro data: When available, incorporate data from assays using human fecal supernatants to estimate degradation rate constants.
- Flag susceptible compounds: Identify candidates with functional groups prone to microbial reduction or hydrolysis (e.g., azo compounds, sulfasalazine analogs) and run scenarios with/without this clearance pathway.
Q3: What are the critical parameters to optimize when scaling IVIVE from microsomes to whole liver for CYP2C9 substrates? A: Focus on these parameters, often refined via Bayesian optimization:
- Fraction unbound in microsomes (fumic): Accurate measurement is critical.
- Microsomal binding correction: Implement a compound-specific binding model.
- Liver-to-plasma partition coefficient (Kp): Use mechanistic methods (e.g., Poulin & Theil, Berezhkovskiy) over simple regression.
- Plasma protein binding (fu): Use human-specific values from relevant in vitro systems.
Experimental Protocols
Protocol 1: Determination of Intrinsic Clearance (CLint) for CYP3A4 Using Human Liver Microsomes (HLM) Objective: To obtain reliable in vitro CLint for scaling to hepatic metabolic clearance. Method:
- Prepare HLM incubation mixtures (0.5 mg/ml protein) in 100 mM phosphate buffer (pH 7.4) with an NADPH-regenerating system.
- Add test compound at a subsaturating concentration (typically 1 µM) and incubate at 37°C.
- Remove aliquots at 0, 5, 10, 20, and 30 minutes, quenching with cold acetonitrile containing internal standard.
- Analyze samples via LC-MS/MS to determine substrate depletion over time.
- Calculate in vitro CLint (µL/min/mg protein) from the substrate depletion rate constant.
- Scale to in vivo hepatic intrinsic clearance using well-stirred model and human liver scaling factors (e.g., 80 mg microsomal protein per gram liver, 25 g liver/kg body weight).
Protocol 2: Parallel Artificial Membrane Permeability Assay (PAMPA) for Predicting Effective Intestinal Permeability (Peff) Objective: To provide a high-throughput, permeability rank-order for passive diffusion. Method:
- Prepare a lipid-dodecane solution (e.g., 2% phosphatidylcholine) to form the artificial membrane on a 96-well filter plate.
- Add donor solution (test compound in pH 6.5 buffer to simulate intestinal pH) to the upper chamber.
- Fill the lower (acceptor) chamber with pH 7.4 buffer.
- Incubate the plate for 4-6 hours at 25°C under gentle agitation.
- Quantify compound concentration in both donor and acceptor wells using a UV plate reader or LC-MS.
- Calculate permeability (Pe, cm/s) and correlate to human in vivo Peff values using a validated in vitro-in vivo correlation (IVIVC).
Data Presentation
Table 1: Impact of Key ADME Parameters on Predicted Oral Bioavailability (F)
| Parameter | Typical Range | Effect on Predicted F | Prioritization Guidance |
|---|---|---|---|
| Effective Intestinal Permeability (Peff) | 0.1 - 20 (x10⁻⁴ cm/s) | Direct, positive correlation. Primary driver for BCS Class I/II compounds. | Prioritize candidates with Peff > 5 x10⁻⁴ cm/s. |
| Hepatic Intrinsic Clearance (CLint,h) | 1 - 1000 (mL/min/kg) | Inverse correlation. Major limiter for high-clearance compounds. | For CYP substrates, target CLint,h < 15 mL/min/kg in human hepatocytes. |
| Fraction Absorbed (Fa) | 0 - 1.0 | Direct, positive correlation. Limits maximum achievable F. | Use PBPK to identify Fa > 0.9. Sensitize to bile salt interactions. |
| Gut Wall Intrinsic Clearance (CLint,gut) | 0 - 500 (µL/min) | Inverse correlation, critical for CYP3A4/UGT1A substrates. | For low F compounds, evaluate CLint,gut contribution > 20% of total first-pass. |
| Blood-to-Plasma Ratio (B/P) | 0.5 - 2.0 | Affects hepatic clearance calculation. High ratio can increase predicted F. | Measure experimentally; do not default to 1.0. |
Table 2: Common IVIVE Scaling Factors for Key Enzymes
| Enzyme System | Scaling Factor (SF) | Physiological Value (Source) | Application Note |
|---|---|---|---|
| CYP3A4 (Liver) | Microsomal Protein per Gram Liver (MPPGL) | 40 - 80 mg/g (Individual variability exists) | Use population distributions, not point estimates. |
| CYP3A4 (Gut) | Enterocyte Protein per cm² | 20 - 40 mg/cm² (Region-specific) | Duodenum/Jejunum primary site; ileum/colon lower abundance. |
| UGT1A1 | Microsomal Protein per Gram Liver | 40 - 80 mg/g | Often co-modeled with CYP3A4 due to overlapping substrates. |
| Hepatic Uptake (OATP1B1) | Hepatocyte Volume per Liver | 1.2 x 10⁹ cells/kg liver | Active uptake can be clearance-rate determining; incorporate plated human hepatocyte data. |
Diagrams
Diagram 1: PBPK Workflow for First-Pass Metabolism Prediction
Diagram 2: Key Pathways Determining Oral Bioavailability
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function & Application in PBPK/IVIVE |
|---|---|
| Pooled Human Liver Microsomes (HLM) | Contains a representative mix of human hepatic CYP enzymes. Used for initial high-throughput intrinsic clearance (CLint) screening and reaction phenotyping. |
| Cryopreserved Human Hepatocytes | Gold-standard for predicting hepatic metabolic clearance and transporter effects. Provides intact cellular machinery for IVIVE scaling. |
| Recombinant CYP Isozymes (rCYP) | Expressed individually in insect or mammalian cells. Critical for identifying the specific enzyme(s) responsible for a compound's metabolism (reaction phenotyping). |
| Transfected Cell Lines (e.g., MDCK, HEK293) | Engineered to express specific human uptake (OATP1B1/1B3) or efflux (P-gp, BCRP) transporters. Used to determine transporter kinetics (Km, Vmax) for mechanistic PBPK. |
| Biorelevant Dissolution Media (FaSSIF/FeSSIF) | Simulates fasted and fed state intestinal fluids. Used in dissolution testing to predict in vivo dissolution and solubility limitations to absorption (Fa). |
| Stable Isotope-Labeled Internal Standards | Essential for accurate and precise quantitation of drug concentrations in complex biological matrices (e.g., microsomal incubations, plasma) via LC-MS/MS. |
| NADPH Regenerating System | Supplies a constant source of NADPH, the essential cofactor for CYP-mediated oxidation reactions, during in vitro metabolic stability assays. |
| Artificial Membranes for PAMPA | Provides a high-throughput, non-cell-based model to assess passive transcellular permeability, a key input for predicting intestinal absorption. |
Current Challenges in Predicting Human First-Pass Effects from Preclinical Data
Troubleshooting Guides & FAQs
FAQ 1: Why do our PBPK models consistently underpredict oral bioavailability for high-clearance compounds when using in vitro metabolic stability data from human liver microsomes (HLM)?
Answer: This is a classic symptom of neglecting non-cytochrome P450 (non-CYP) pathways and gut wall metabolism. HLM primarily contain CYP enzymes but are deficient in many Phase II conjugating enzymes (e.g., UGTs) and extrahepatic enzymes. Furthermore, first-pass extraction occurs in both the gut and liver.
Troubleshooting Guide:
- Check Enzyme Coverage: Validate your in vitro system. For compounds suspected of undergoing glucuronidation, sulfation, or hydrolysis, supplement HLM studies with human liver S9 fractions or recombinant enzymes.
- Incorporate Gut Metabolism: Integrate data from human intestinal microsomes or consider using a permeability-limited gut model (e.g., the Advanced Dissolution, Absorption, and Metabolism - ADAM model) within your PBPK software.
- Verify Input Parameters: Ensure the correct scaling factors (e.g., microsomal protein per gram of liver, MPPGL) and physiological values (intestinal blood flow, villous surface area) are used.
FAQ 2: How should we handle significant species differences in enzyme affinity (Km) when scaling from rat to human?
Answer: Direct scaling using rat in vivo clearance data with humanized Km values is error-prone. The recommended approach is to use in vitro human enzyme kinetic data whenever possible.
Troubleshooting Guide:
- Conduct In Vitro Kinetics: Perform Michaelis-Menten kinetics in human hepatocytes or recombinant human enzymes to obtain intrinsic clearance (CLint) and Km.
- Apply Relative Activity Factor (RAF): If using recombinant enzymes, apply a RAF to scale activity to physiologically relevant levels.
- Use Species-Specific PBPK: Build a verified rat PBPK model using rat-specific Km and physiology to validate the in vitro-in vivo extrapolation (IVIVE) approach. Then, build the human model using human in vitro parameters and physiology.
FAQ 3: Our model fails when a drug shows pH-dependent solubility and is a substrate for efflux transporters (e.g., P-gp). How do we parameterize this complex interaction?
Answer: This requires a mechanistic, dynamic model that accounts for changing luminal conditions and transporter saturation along the gastrointestinal tract.
Troubleshooting Guide:
- Characterize Solubility & Permeability: Measure solubility across a physiologically relevant pH range (1.5 to 7.5). Determine apparent permeability (Papp) in Caco-2 or MDCK assays with and without a potent P-gp inhibitor (e.g., zosuquidar).
- Obtain Transporter Kinetic Parameters: If possible, determine the Michaelis-Menten constants (Km and Vmax) for the efflux transporter using transfected cell systems.
- Select Appropriate Model Structure: Use a PBPK platform that supports compartmental gut models with integrated pH-dependent dissolution and saturable, region-specific transporter expression.
Experimental Protocols for Key Assays
Protocol 1: Determining Fraction Metabolized (fm) by Different Pathways Using Chemical Inhibitors in Human Hepatocytes
Objective: To quantify the fractional contribution of specific enzymes (e.g., CYP3A4) to the overall hepatic metabolism of a drug candidate.
Methodology:
- Incubation Setup: Prepare suspensions of cryopreserved human hepatocytes (≥1 million cells/mL) in incubation medium.
- Pre-incubation: Add a selective chemical inhibitor (e.g., 1 µM ketoconazole for CYP3A4, 10 µM quinidine for CYP2D6) or vehicle control. Pre-incubate for 15 minutes at 37°C.
- Reaction Initiation: Add the test compound at a concentration ≤ Km. Inculate for a predetermined linear time (e.g., 30-60 minutes).
- Reaction Termination: Quench with an equal volume of acetonitrile containing internal standard.
- Analysis: Quantify parent compound loss using LC-MS/MS.
- Calculation: Calculate the remaining fraction of parent drug in inhibited vs. control incubations. The fm for the inhibited pathway is: fm = 1 - (Amountparent with inhibitor / Amountparent control).
Protocol 2: Simultaneous Assessment of Metabolic Stability and Transporter Efflux in a Single System (Caco-2/MDCK-MDR1 cells)
Objective: To obtain integrated parameters for gut permeability, efflux, and intestinal metabolism.
Methodology:
- Cell Culture: Seed Caco-2 or MDCK-MDR1 cells on transwell inserts and culture for 21 days (Caco-2) or 7 days (MDCK) to form confluent, differentiated monolayers.
- Bidirectional Transport Assay: Add test compound to the donor compartment (apical, A, or basolateral, B) and blank buffer to the receiver compartment. Incubate at 37°C.
- Sampling: Take samples from the receiver compartment at regular intervals (e.g., 30, 60, 90, 120 min). Also sample the donor compartment at start and end.
- Inhibition Arm: Run parallel experiments with a potent P-gp/BCRP inhibitor in both compartments.
- Metabolite Screening: Analyze receiver and donor samples using LC-HRMS to identify and quantify any metabolites formed during transit.
- Data Analysis:
- Calculate apparent permeability (Papp).
- Determine efflux ratio (ER) = Papp(B→A) / Papp(A→B).
- Calculate fraction metabolized during transport.
Data Presentation
Table 1: Common In Vitro Systems for First-Pass Metabolism Parameter Generation
| System | Primary Use | Key Strengths | Key Limitations | Typical Output for PBPK |
|---|---|---|---|---|
| HLM/S9 | Hepatic CLint, Reaction phenotyping | High throughput, low cost, minimal lot variation | Lack full cellular context, may miss non-CYP enzymes | Unbound CLint (µL/min/mg protein) |
| Human Hepatocytes | Hepatic CLint, non-CYP metabolism, transporter interplay | Full complement of hepatic enzymes & cofactors, physiological | Donor variability, lower throughput, cost | Unbound CLint (µL/min/10^6 cells) |
| Recombinant Enzymes | Reaction phenotyping, enzyme kinetics | Pure system for specific enzymes | Non-physiological expression levels, no enzyme interplay | Vmax & Km |
| Intestinal Microsomes | Gut wall metabolism | Direct assessment of intestinal CYP/UGT activity | No transporter activity, no absorption component | Gut wall CLint |
| Caco-2 Cells | Permeability, efflux, gut metabolism | Integrated system for absorption & metabolism | Variable expression levels, long culture time | Papp, Efflux Ratio, fm_gut |
Table 2: Quantitative Impact of Common Oversights on Predicted Oral Bioavailability (F)
| Oversight in Preclinical Data | Typical Error in Predicted Human F | Mechanism |
|---|---|---|
| Ignoring UGT-mediated metabolism | Overprediction by 20-50% for some compounds | Missed significant Phase II first-pass extraction |
| Using hepatic data only for a high gut-extraction drug | Overprediction by 30-70% | Neglects first-pass loss in enterocytes |
| Applying in vivo rodent fm without correction | Unpredictable; can be over- or under-prediction | Species differences in enzyme abundance/affinity |
| Not accounting for plasma protein binding in IVIVE | Underprediction for high-bound, low-clearance drugs | Incorrect estimation of free drug available for metabolism |
Visualizations
Title: PBPK Modeling Workflow & Challenge Identification
Title: First-Pass Extraction Sites: Gut and Liver
The Scientist's Toolkit: Research Reagent Solutions
| Item / Reagent | Function in First-Pass Research | Key Consideration |
|---|---|---|
| Cryopreserved Human Hepatocytes | Gold standard for measuring intrinsic hepatic clearance & metabolite identification. | Pooled donors reduce variability; check viability & activity certificates. |
| Selective Chemical Inhibitors (e.g., Ketoconazole, Quinidine, BNPP) | To determine fraction metabolized (fm) by specific enzyme pathways in hepatocytes or microsomes. | Use at recommended, selective concentrations to avoid off-target inhibition. |
| Transfected Cell Lines (e.g., MDCK-MDR1, HEK-UGT1A1) | Isolate contribution of specific transporters or enzymes to permeability/metabolism. | Compare to wild-type controls to assess background activity. |
| Biorelevant Media (FaSSIF/FeSSIF) | Simulate intestinal fluids for solubility and dissolution testing under physiological conditions. | Critical for accurately modeling absorption of poorly soluble compounds. |
| Stable Isotope-Labeled Drug | Used as an internal standard in complex in vitro systems to track parent loss and metabolite formation. | Essential for accurate LC-MS/MS quantification in biological matrices. |
| PBPK Software Platform (e.g., GastroPlus, Simcyp, PK-Sim) | Integrate all preclinical data to build mechanistic models and simulate human PK. | Choose based on model flexibility, built-in populations, and regulatory acceptance. |
Building and Applying PBPK Models for First-Pass Metabolism Prediction
Technical Support Center: Troubleshooting & FAQs
FAQ 1: My model consistently under-predicts in vivo hepatic clearance compared to observed clinical data. What are the primary sources of this discrepancy?
- Answer: Under-prediction often stems from incomplete characterization of metabolic processes. Key troubleshooting steps include:
- Verify Enzyme Kinetic Parameters: Ensure your in vitro
VmaxandKmare scaled appropriately using accurate ISEF (Inter-System Extrapolation Factor) values for your specific recombinant enzyme system or hepatocyte batch. Generic scaling factors may not apply. - Check for Non-Specific Binding: Neglecting non-specific binding in in vitro incubations (
fu_inc) can lead to an overestimation of intrinsic clearance. Re-measurefu_incand incorporate it into your in vitro-in vivo extrapolation (IVIVE). - Consider Extrahepatic Metabolism: Review literature for evidence of gut wall or renal metabolism. For orally administered drugs, integrate gut
ft(fraction transported) andfg(fraction escaping gut metabolism) into your PBPK model's first-pass prediction. - Evaluate Transporter Effects: Hepatic uptake (e.g., via OATP1B1/1B3) can significantly influence clearance. If relevant, incorporate uptake
CLintinto your liver model.
- Verify Enzyme Kinetic Parameters: Ensure your in vitro
FAQ 2: How do I properly incorporate plasma protein binding and blood-to-plasma ratio data into my PBPK model for accurate first-pass prediction?
- Answer: These parameters are critical for partitioning. Use the following structured approach:
- Measure or source accurate fraction unbound in plasma (
fu_p) and blood-to-plasma concentration ratio (Cb/Cp). - In your PBPK software, set the compound's Fraction Unbound in Plasma (
fu_p) and Blood-to-Plasma Ratio (BPR) as direct inputs. - The model will use these to calculate the fraction unbound in blood (
fu_b) and the effective concentration available for hepatic enzymes. Incorrect input here can skew predicted hepatic extraction.
- Measure or source accurate fraction unbound in plasma (
FAQ 3: The predicted AUC after oral administration is inaccurate despite good IV prediction. What should I focus on for first-pass metabolism?
- Answer: This points to an error in modeling the pre-systemic extraction pathway. Focus on the gut-liver axis:
- Gut Metabolism: Confirm the value for the fraction escaping gut metabolism (
fg). This is often derived from in vitro data using human intestinal microsomes or recombinantly expressed CYP3A4, coupled with appropriate scaling models. - Hepatic Availability: Re-evaluate the hepatic extraction ratio (
EH) calculation. Ensure the liver model correctly uses the well-stirred, parallel-tube, or dispersion model as appropriate for your compound. - Absorption & Solubility: Poor predicted absorption due to incorrect solubility or permeability inputs can also affect AUC. Verify these physicochemical parameters.
- Gut Metabolism: Confirm the value for the fraction escaping gut metabolism (
Data Presentation: Key In Vitro Parameters for IVIVE
Table 1: Essential In Vitro Parameters for Hepatic Clearance IVIVE
| Parameter | Symbol | Typical Experiment | Purpose in PBPK Model |
|---|---|---|---|
| Michaelis Constant | Km |
Microsomal/ Hepatocyte Incubation | Defines enzyme-substrate affinity. Used to scale in vitro CLint. |
| Maximum Velocity | Vmax |
Microsomal/ Hepatocyte Incubation | Defines maximal metabolic rate. Scaled per gram of liver. |
| Fraction Unbound in Incubation | fu_inc |
Equilibrium Dialysis/ Ultracentrifugation | Corrects in vitro CLint for non-specific binding in assay. |
| Intrinsic Clearance | CLint,in vitro |
Substrate Depletion or Metabolite Formation | Direct input or derived from Vmax/Km. Basis for IVIVE. |
| Inter-System Extrapolation Factor | ISEF | Comparative Activity Assessment | Corrects for activity differences between recombinant enzymes and human tissue. |
| Fraction Unbound in Plasma | fu_p |
Equilibrium Dialysis/ Ultracentrifugation | Determines free drug concentration for hepatic clearance and tissue partitioning. |
Table 2: Key First-Pass Metabolism Parameters
| Parameter | Symbol | Source/Calculation | Impact on Oral Bioavailability (F) |
|---|---|---|---|
| Fraction Absorbed | Fa |
In vitro permeability (e.g., Caco-2, PAMPA) | F = Fa * Fg * Fh. Direct multiplier. |
| Fraction Escaping Gut Metabolism | Fg |
In vitro intestinal microsome data + Qgut model | Critical for CYP3A4/CYP2D6 substrates. |
| Hepatic Availability | Fh |
Fh = 1 - EH where EH from scaled CLint |
Determined by hepatic blood flow (Qh) and free CLint. |
Experimental Protocols
Protocol 1: Determination of Intrinsic Clearance (CLint) via Substrate Depletion in Human Liver Microsomes (HLM)
- Incubation Setup: Prepare HLM (e.g., 0.5 mg/mL protein) in 100 mM phosphate buffer (pH 7.4). Pre-warm at 37°C.
- Reaction Initiation: Add a low, non-saturating concentration of test compound (typically <<
Km, e.g., 1 µM). Initiate reaction by adding NADPH-regenerating system. - Time Course Sampling: Aliquot the incubation mixture at multiple time points (e.g., 0, 3, 7, 15, 30, 45 min) and immediately quench with an equal volume of acetonitrile containing internal standard.
- Analysis: Centrifuge quenched samples, analyze supernatant via LC-MS/MS to determine parent compound depletion over time.
- Calculation: Fit the natural log of percentage remaining vs. time data to a first-order decay model. The slope is the in vitro depletion rate constant (
k_depl). CalculateCLint, in vitro = k_depl / [microsomal protein concentration].
Protocol 2: Measurement of Fraction Unbound in Incubation (fu_inc)
- Setup: Use a 96-well equilibrium dialysis device. Load one side (donor) with incubation matrix (HLM in buffer at typical assay concentration) spiked with test compound.
- Dialyze: Load the other side (receiver) with blank buffer. Seal plate and incubate at 37°C with gentle agitation for 4-6 hours to reach equilibrium.
- Post-Dialysis Analysis: Sample from both donor and receiver compartments. Analyze concentrations using LC-MS/MS.
- Calculation:
fu_inc = [Concentration in Receiver] / [Concentration in Donor] at equilibrium.
Mandatory Visualization
PBPK First-Pass Prediction Workflow
First-Pass Metabolism Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for In Vitro-In Vivo Extrapolation (IVIVE)
| Item | Function in PBPK Research |
|---|---|
| Human Liver Microsomes (HLM) | Pooled donor preparation containing membrane-bound Phase I/II enzymes for intrinsic clearance (CLint) assays. |
| Recombinant Human CYPs (rCYPs) | Individual cytochrome P450 isoforms expressed in insect cells for reaction phenotyping and obtaining isoform-specific kinetics. |
| NADPH Regenerating System | Provides a constant supply of NADPH, the essential cofactor for CYP450-mediated oxidation reactions. |
| Cryopreserved Human Hepatocytes | Gold-standard in vitro system containing full complement of hepatic enzymes and transporters for more holistic clearance assessment. |
| Equilibrium Dialysis Device | Standard method for determining fraction unbound (fu_inc, fu_p) via passive diffusion equilibrium across a semi-permeable membrane. |
| LC-MS/MS System | High-sensitivity analytical platform for quantifying low concentrations of drug and metabolites in complex biological matrices. |
| PBPK Software Platform | Simulation environment (e.g., GastroPlus, Simcyp, PK-Sim) with built-in physiological databases to implement IVIVE and run PBPK models. |
Technical Support Center: Troubleshooting Guides & FAQs for PBPK First-Pass Metabolism
FAQ 1: How do I determine the most appropriate enzyme abundance values (e.g., CYP3A4) for my human liver PBPK model?
Answer: The selection of enzyme abundance values is a critical step. Common issues arise from using values from incompatible sources (e.g., mixing in vitro pmol/mg protein with in vivo pmol/g tissue). Recent consortia have published standardized values.
- Solution: Use consensus values from recent literature. For example, the International Transporter Consortium and other groups have compiled in vivo relevant abundances. Always ensure the units are consistent with your model's tissue composition definitions (per gram of tissue vs. per mg microsomal protein). Convert using measured or estimated hepatocellularity and microsomal protein per gram of liver (typical: 99 million hepatocytes/g liver, 40 mg microsomal protein/g liver).
Data Table: Example Consensus CYP Enzyme Abundance in Human Liver
| Enzyme | Abundance (pmol/mg microsomal protein) | Abundance (pmol/g liver) | Key Source / Notes |
|---|---|---|---|
| CYP3A4 | 82 - 137 | 3280 - 5480 | Achour et al., 2014; Barter et al., 2007 |
| CYP2D6 | 8 - 15 | 320 - 600 | Polymorphic, major source of variability. |
| CYP2C9 | 69 - 110 | 2760 - 4400 | Use genotype-specific values if available. |
| CYP1A2 | 34 - 52 | 1360 - 2080 | Inducible; consider smoking status. |
FAQ 2: My model consistently under-predicts intestinal first-pass metabolism. What are the key inputs I might be missing?
Answer: Under-prediction of gut wall metabolism often stems from oversimplified inputs.
- Regional Variation: CYP3A4 and UGT abundance is not uniform along the gastrointestinal tract. The duodenum and jejunum have much higher expression than the colon.
- Villus Blood Flow: The effective blood flow delivering drug to enterocytes is a fraction of the total splanchnic blood flow. Incorrect fractional flow will skew extraction predictions.
- Transporter Interplay: For substrates of efflux transporters like P-gp, the sequential metabolism and efflux (enterocyte cycling) is crucial. Ensure your model structure captures this interplay.
Experimental Protocol: Determining Regional Intestinal Enzyme Abundance
- Method: Use human intestinal samples (from organ donor or surgical resections) categorized by region: duodenum, jejunum, ileum, colon.
- Sample Prep: Homogenize mucosal scrapings. Prepare microsomes or S9 fractions via differential centrifugation.
- Quantification: Use quantitative targeted proteomics (e.g., LC-MS/MS with peptide standards) to measure absolute abundance of specific enzymes (CYP3A4, UGTs). Normalize data per mg of total protein or per cm of intestinal length.
- Data Integration: Map abundances to corresponding intestinal segment lengths and radii in the PBPK model.
FAQ 3: How should I incorporate variable tissue composition (e.g., fractional volumes of blood, water, lipid) for different populations?
Answer: Tissue composition directly affects drug partitioning. Using default values for a 70kg male will introduce errors for special populations.
- Solution: Implement age- or population-specific tissue composition tables. Key resources include:
- ICRP Publications: Reference values for the male and female adult.
- Pediatric & Geriatric Data: Use published models that estimate water, fat, and protein content changes with age.
- Disease States: For conditions like obesity or cirrhosis, literature values for altered organ cellularity and fat content must be sourced.
- Action: Always run a sensitivity analysis on tissue composition parameters to understand their impact on your model's predicted plasma and tissue concentration-time profiles.
Research Reagent Solutions Toolkit
| Item | Function in PBPK-Related Research |
|---|---|
| Quantitative Proteomics Kits (e.g., SIL peptide standards for CYPs/UGTs) | Absolute quantification of enzyme and transporter abundances in human tissue samples. |
| Pooled Human Liver Microsomes (HLM) & Hepatocytes | In vitro system for measuring intrinsic clearance and scaling to in vivo. |
| Recombinant Human Enzymes (rCYP, rUGT) | Reaction phenotyping to identify enzymes responsible for metabolism. |
| Physiologically Relevant Buffer Systems (e.g., FaSSIF/FeSSIF) | For assessing solubility and dissolution in gut lumen for oral absorption modeling. |
| PBPK Software Platforms (e.g., GastroPlus, Simcyp, PK-Sim) | Contain built-in databases for physiology, enzyme abundances, and trial design. |
Diagram 1: Key Inputs for a Liver PBPK First-Pass Model
Diagram 2: Troubleshooting Under-Prediction of Gut Metabolism
Technical Support Center: Troubleshooting & FAQs
This support center addresses common issues encountered when using leading PBPK platforms in the context of predict-first first-pass metabolism research, as framed by our broader thesis.
Frequently Asked Questions
Q1: In GastroPlus, my simulated hepatic bioavailability (Fh) is consistently overestimated for CYP3A4 substrates, despite accurate in vitro CLint data. What could be the cause? A: This often stems from improper scaling of the in vitro-to-in vivo intrinsic clearance (CLint). A primary troubleshooting step is to verify the "Periportal Binding" and "In Vitro Binding" settings. Ensure the in vitro binding correction matches your assay conditions (e.g., microsomal protein concentration). For CYP3A4, consider enabling the "Gut Metabolism" module even for oral dosing, as intestinal extraction may be significant. Re-check the IVIVE method (e.g., Rodgers & Rowland vs. conventional) selected in the Compound > Metabolism tab.
Q2: Simcyp Simulator reports "Inability to achieve target AUC" during a Population Simulator run for a drug with high first-pass effect. How should I proceed? A: This error typically relates to the optimization algorithm failing with your current parameter bounds. Follow this protocol:
- Isolate the Issue: Run a single "Mean Subject" simulation first to ensure the base model works.
- Adjust Bounds: In the Trial Design pane, navigate to the Dosing Regimen section. Expand "Advanced Options" and increase the "Upper Limit" for the dose search (e.g., from default 100 mg to 1000 mg) if the first-pass effect is very high.
- Check Enzyme Abundance: Verify that the population-specific enzyme abundance (e.g., CYP2D6 in your selected population) is not set to zero or an extreme value in the Population tab.
Q3: PK-Sim generates unexpected, very low plasma concentrations for an orally administered compound with known solubility limitations. Which parameters are most critical to review? A: This points to potential mis-specification of dissolution or solubility. Use this checklist:
- Solubility Table: Confirm solubility is entered at the correct pH values (especially gastric and intestinal pH). Use the "Solubility at pH" table, not a single value.
- Dissolution Model: In the "Formulation" properties, switch from "Default" to the "Dissolution Model" and select an appropriate model (e.g.,
Weibull). Adjust the time parameters to reflect your dissolution data. - Particle Size: In the "Compound Properties" > "Distribution", ensure the "Mean particle radius" is realistically set (typically 25-50 µm for a standard formulation, not the default 1 µm).
Key Experiment Protocol: IVIVE of Hepatic Clearance for PBPK Model Qualification
Objective: To generate in vivo pharmacokinetic predictions from in vitro metabolism data for the purpose of qualifying a PBPK platform's first-pass metabolism prediction capability.
Detailed Methodology:
- In Vitro Data Input: Obtain intrinsic clearance (CLint) from human liver microsomes (HLM) or hepatocytes for the compound of interest.
- Data Normalization: Normalize CLint values per million hepatocytes or mg microsomal protein.
- Platform-Specific IVIVE Setup:
- GastroPlus (Metabolism & Transport Module): Navigate to
Compound > Metabolism. Input the CLint value. Select the appropriateIVIVE Method(e.g., "Traditional", "Rodgers & Rowland"). Input thefu,inc(fraction unbound in incubation). - Simcyp (Compound Model): In the
Compoundfile, underEnzyme Kinetics, inputCLintandfu,inc. Select the relevantEnzymeand its abundance value. Choose the desiredIVIVEmethod from the "Physiological Models" (e.g.,Sim-AllometricorSim-Population). - PK-Sim (Ontogeny & Variation): In the
IndividualorPopulationbuilding blocks, assign the process"Hepatic Clearance via Specific Enzyme"to the compound. Input theSpecific clearancederived from in vitro data. Define theOntogenyprofile for the enzyme if simulating varied populations.
- GastroPlus (Metabolism & Transport Module): Navigate to
- Simulation Execution: Run a single IV bolus or oral administration simulation in a "Virtual Healthy Volunteer" population.
- Model Qualification: Compare the simulated plasma concentration-time profile and derived parameters (AUC, CL, Fh) against observed clinical data from a single oral dose study. Use diagnostic plots (observed vs. predicted) and fold-error analysis (acceptable range: 0.5 - 2.0).
Data Presentation: Comparison of Leading PBPK Platform Capabilities
Table 1: Core Capabilities for First-Pass Metabolism Prediction
| Feature / Capability | GastroPlus (v9.9+) | Simcyp Simulator (v22+) | PK-Sim / MoBi (v11+) |
|---|---|---|---|
| Primary IVIVE Method | Traditional, Rodgers & Rowland, MI | Regression- and mechanistic-based (RAF, ISEF, POP) | Standard organ-blood clearance model, extended to cellular level |
| Gut Wall Metabolism | Advanced Compartmental Absorption & Transit (ACAT) model with gut metabolism | Full GI tract model with enterocyte-level metabolism | Intestinal segment model with enzyme expression |
| Enzyme Database | Built-in, user-expandable | Extensive, pre-loaded (SNP, abundance, ontogeny) | User-defined, with import functionality |
| CYP Inhibition Modeling | Competitive, uncompetitive, time-dependent (TDI) | Mechanistic, static & dynamic (DDI) | Competitive, mechanism-based (MBI) |
| Population Simulation | Built-in demographics, limited genetic polymorphism | Highly advanced, genotype-driven populations | Flexible, based on parameter distributions |
| Key First-Pass Output | Fh, FaFg, Qgut | Fh, Fg, organ extraction ratios | Hepatic extraction ratio, systemic clearance |
Table 2: Common Troubleshooting Targets by Platform
| Issue Symptom | Likely Parameter (GastroPlus) | Likely Parameter (Simcyp) | Likely Parameter (PK-Sim) |
|---|---|---|---|
| Overestimated Oral AUC | f<sub>u,inc</sub>, Periportal Binding Factor |
ISEF/RAF Value, Enterocyte Blood Flow |
Intrinsic Clearance, K<sub>m</sub> Value |
| Underestimated Cmax | Dissolution Rate, Particle Radius |
Transit Rate (GI Model), Peff |
Solubility Table, Lag Time |
| Poor IV/PO Concordance | First-Pass Organ Selection (Lung vs. Liver) |
Route of Administration specific model selection |
Administration Protocol (Application Type) |
Visualizations
Diagram 1: PBPK First-Pass Metabolism Prediction Workflow
Diagram 2: Key Interactions in Gut-Liver First-Pass Axis
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagent Solutions for Supporting In Vitro First-Pass Metabolism Assays
| Item | Function in PBPK Context | Typical Vendor/Example |
|---|---|---|
| Human Liver Microsomes (HLM) | Source of cytochrome P450 enzymes for measuring intrinsic clearance (CLint) and kinetic parameters (Km, Vmax). | Corning Life Sciences, Xenotech |
| Cryopreserved Human Hepatocytes | Integrated cellular system to study phase I/II metabolism, transporter effects, and provide a more physiologically relevant CLint. | BioIVT, Lonza |
| Specific CYP Isoform Inhibitors (e.g., Ketoconazole-CYP3A4) | To verify the enzyme responsible for metabolism and deconvolute contributions in HLM assays. | Sigma-Aldrich, Cayman Chemical |
| NADPH Regenerating System | Essential cofactor system to sustain CYP450 activity during in vitro metabolic stability incubations. | Promega, Thermo Fisher Scientific |
| Dialysis Membranes / Charcoal | For determining fraction unbound in incubation (fu,inc), a critical parameter for accurate IVIVE. | Harvard Apparatus, Sigma-Aldrich |
| LC-MS/MS Grade Solvents & Standards | For high-sensitivity quantification of substrate depletion or metabolite formation in in vitro assays. | Fisher Chemical, Sigma-Aldrich |
This technical support center provides troubleshooting guidance and FAQs for researchers conducting PBPK (Physiologically Based Pharmacokinetic) modeling to predict the bioavailability of high-extraction ratio drugs, a critical component of thesis work focused on first-pass metabolism prediction.
FAQs & Troubleshooting
Q1: My PBPK model consistently overpredicts the oral bioavailability (F) of a high-extraction ratio drug. What are the primary parameters to investigate? A: This is a common calibration challenge. Prioritize investigating:
- Intestinal and Hepatic Blood Flow Rates: Ensure values are physiologically accurate for your species (e.g., human, rat). Slight overestimations can significantly under-predict first-pass extraction.
- Intrinsic Clearance (CLint) Input: Verify the in vitro-in vivo extrapolation (IVIVE) of metabolic CLint. Check the scaling factors (e.g., microsomal protein per gram of liver, hepatocellularity) and the assumption of non-restrictive clearance. For high-extraction drugs, bioavailability is highly sensitive to CLint.
- Enterocyte Metabolism: For drugs metabolized by CYP3A4 or UGTs, incorporate gut wall metabolism (Fg) using appropriate enzyme abundance data. Omitting this can lead to overpredictions of F.
- Plasma Protein Binding: Confirm the accuracy of the fraction unbound (fu) input, especially if measured under different conditions (e.g., pH, temperature).
Q2: During IVIVE, what are the critical considerations for scaling enzyme kinetic data (Vmax, Km) from recombinant systems to whole organ intrinsic clearance? A: Key considerations include:
- Enzyme Abundance Scaling: Use tissue-specific abundance data (pmol enzyme per mg microsomal protein or per gram of tissue) to scale from recombinant systems.
- Relative Activity Factor (RAF): Apply RAFs to account for differences in catalytic activity between recombinant enzymes and human tissue microsomes.
- Nonspecific Binding in In Vitro Assays: Correct the apparent Km for nonspecific binding to in vitro assay components (e.g., microsomes, plastic) to obtain the unbound Km (Km,u), which is critical for accurate IVIVE.
- Inter-system Extrapolation Factors (ISEF): For CYP enzymes, consider applying isoform-specific ISEFs to bridge the activity gap between recombinant systems and native human liver microsomes.
Q3: How should I handle transporter-mediated hepatic uptake for a high-extraction drug where clearance appears perfusion-rate limited? A: Even for perfusion-limited drugs, transporter kinetics can influence intracellular concentration at the enzyme site. To troubleshoot:
- Sensitivity Analysis: Perform a local sensitivity analysis on the hepatic uptake clearance parameter. If F is sensitive to this parameter, more rigorous characterization is needed.
- Incorporating Transport: Implement a permeability-limited or dispersion liver model (e.g., full PBPK or "minimal PBPK") instead of a simple well-stirred liver model. This allows separate definition of sinusoidal uptake and efflux clearances.
- Data Requirement: You may need in vitro transporter data (e.g., HEK293 cells overexpressing OATP1B1/1B3) to inform the uptake clearance parameter. Without data, consider if the drug is a known substrate from literature.
Q4: What experimental protocol is recommended for validating a PBPK model's prediction of first-pass metabolism? A: A robust validation protocol involves multiple, complementary study designs:
- Human Pharmacokinetic (PK) Study: Conduct a crossover study in healthy volunteers with simultaneous intravenous (IV) and oral administration of the drug.
- Bioanalysis: Use a validated LC-MS/MS method to quantify parent drug (and major metabolites, if possible) in plasma samples collected over an appropriate time period.
- Data Analysis: Calculate the observed absolute bioavailability: Fobs = (AUCpo * Doseiv) / (AUCiv * Dose_po).
- Model Validation: Compare the PBPK-predicted F, AUC_po, and Cmax with the observed values. Successful prediction is typically within a 2-fold error range, though for high-extraction drugs, a tighter criterion (e.g., 1.5-fold) for F is ideal. Visual predictive checks and comparison of predicted vs. observed concentration-time profiles are essential.
Table 1: Key Physicochemical and Pharmacokinetic Parameters for High-Extraction Ratio Model Drugs
| Parameter | Propranolol (Example) | Midazolam (Example) | Alprenolol (Example) | Notes |
|---|---|---|---|---|
| Log P | 3.21 | 3.83 | 3.10 | High lipophilicity facilitates membrane diffusion and enzyme access. |
| fu (Fraction Unbound) | 0.15 | 0.03 | 0.20 | High protein binding reduces free drug concentration for metabolism. |
| Blood-to-Plasma Ratio | 0.95 | 0.70 | 0.85 | Important for converting plasma clearance to blood clearance. |
| Primary Metabolizing Enzyme | CYP2D6, CYP1A2 | CYP3A4/5 | CYP2D6, CYP2C8 | Defines the IVIVE and enzyme abundance scaling path. |
| Hepatic CLint (µL/min/million cells) | ~2500 | ~5000 | ~3000 | High in vitro intrinsic clearance is a hallmark. |
| Reported Human Bioavailability (F%) | ~25% | ~30% | ~10% | Low F due to significant first-pass extraction. |
Table 2: Common IVIVE Scaling Factors for Human Liver
| Scaling Factor | Typical Value | Unit | Function in Calculation |
|---|---|---|---|
| Microsomal Protein per Gram Liver (MPPGL) | 45 | mg/g | Scales microsomal CLint to whole liver CLint. |
| Liver Weight | 20 | g/kg bw | Converts per gram liver values to whole organ. |
| Hepatocellularity | 110 | million cells/g liver | Scales cellular CLint (from hepatocytes) to whole liver CLint. |
Experimental Protocols
Protocol 1: Determination of Intrinsic Clearance (CLint) using Human Hepatocytes Objective: To obtain in vitro metabolic clearance data for IVIVE. Method:
- Incubation: Incate human cryopreserved hepatocytes (0.5-1.0 million cells/mL) with the test drug (≤1 µM) in a suitable medium (e.g., Williams' E) at 37°C under 5% CO2.
- Sampling: At predetermined time points (e.g., 0, 5, 15, 30, 60, 90 min), remove aliquots and immediately quench with an equal volume of acetonitrile containing internal standard.
- Analysis: Centrifuge, collect supernatant, and analyze parent drug concentration using LC-MS/MS.
- Calculation: Fit the natural log of remaining parent concentration vs. time. The slope is the elimination rate constant (k, min⁻¹). CLint (µL/min/million cells) = k / (cell density in million cells/mL).
Protocol 2: Parallel Artificial Membrane Permeability Assay (PAMPA) for Effective Permeability (Peff) Objective: To estimate passive transcellular permeability, a key input for absorption in PBPK models. Method:
- Plate Preparation: Use a PAMPA plate system. Add donor solution (drug in pH 7.4 buffer) to the donor well.
- Membrane Formation: Add a lipid-infused membrane (e.g., lecithin in dodecane) to the filter.
- Assay: Place acceptor plate (pH 7.4 buffer with sink conditions) on top. Incubate at room temperature for 2-6 hours.
- Analysis: Quantify drug in both donor and acceptor compartments via UV or LC-MS. Calculate Peff (cm/s) using a standardized equation that accounts for membrane area, incubation time, and concentration gradient.
Visualizations
PBPK Model Workflow for Predicting Bioavailability
First-Pass Metabolism Pathways (Gut & Liver)
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in PBPK/First-Pass Research |
|---|---|
| Cryopreserved Human Hepatocytes | Gold-standard in vitro system for measuring hepatic metabolic intrinsic clearance (CLint) and performing IVIVE. |
| Recombinant CYP Enzymes | Used to identify the specific cytochrome P450 isoforms responsible for metabolism and to obtain enzyme kinetic parameters (Vmax, Km). |
| Transfected Cell Lines (e.g., OATP-HEK293) | Used to characterize hepatic uptake transporter kinetics, a critical parameter for permeability-limited PBPK models. |
| Human Liver Microsomes (HLM) | A cost-effective system for measuring metabolic stability and reaction phenotyping via chemical inhibitors. |
| PAMPA Kit | High-throughput method for estimating passive transcellular permeability (Peff), a key input for GI absorption models. |
| Rapid Equilibrium Dialysis (RED) Device | Standard method for determining plasma protein binding (fraction unbound, fu) under physiological conditions. |
| PBPK Software Platform (e.g., Simcyp, GastroPlus, PK-Sim) | Industry-standard platforms containing pre-built physiological models, databases (enzyme abundances, demographics), and IVIVE tools. |
| Stable Isotope-Labeled Internal Standards | Critical for accurate and precise LC-MS/MS bioanalysis of drugs and metabolites in complex biological matrices. |
Troubleshooting Guides & FAQs for PBPK Modeling of First-Pass Metabolism
Q1: Our PBPK model consistently underestimates the oral bioavailability of a prodrug. What are the primary formulation-related factors to investigate?
A: Underestimation often stems from incomplete model parameterization of the formulation's behavior. Key factors to check:
- Dissolution Rate: The in vitro dissolution profile may not reflect the in vivo conditions (pH, agitation). Ensure biorelevant media (e.g., FaSSIF/FeSSIF) are used for dissolution testing.
- Pre-systemic Metabolism Location: The model may default to hepatic metabolism only. For prodrugs designed to target intestinal enzymes, verify that gut wall metabolism (
KgutorCLint,gut) is accurately populated, often requiring data from intestinal S9 fractions or Caco-2 cell models. - Carrier-Mediated Transport: If the prodrug or its active moiety utilizes influx/efflux transporters (e.g., PEPT1, P-gp), these kinetic parameters (
Km,Vmax,Jmax) must be incorporated into the gut lumen and enterocyte compartments.
Q2: How can we troubleshoot discrepancies between predicted and observed plasma concentration-time profiles for a targeted drug delivery system (e.g., nanoparticles) when modeling hepatic first-pass?
A: Discrepancies, especially in the absorption phase and early time points, often relate to the release and uptake mechanisms.
- Release Kinetics: The "release" sub-model (e.g., zero-order, Higuchi, or a more complex mechanistic erosion model) must be validated against in vivo relevant trigger data (e.g., pH-dependent release in different GI segments).
- Uptake Mechanism: The default passive diffusion for the API may not apply. For nanoparticles, you must model a distinct "nanoparticle" species with its own uptake rate (e.g., via M-cells or enterocyte endocytosis) and a separate intracellular release step. This requires separate rate constants.
- Organ-Specific Distribution: Verify that the tissue partition coefficients (e.g., Kp) for the delivery system or the released drug in organs like the liver and spleen are informed by biodistribution studies.
Q3: When parameterizing a PBPK model for a prodrug, what is the best approach to obtain reliable intrinsic clearance values for both the prodrug and the active metabolite?
A: A sequential in vitro to in vivo extrapolation (IVIVE) approach is critical.
- Use Appropriate Enzyme Sources: For hepatic clearance, use human liver microsomes (HLM) or hepatocytes. For gut wall metabolism, use human intestinal microsomes (HIM) or S9 fractions.
- Design Incubations Properly: Conduct separate incubations for the prodrug (to measure its direct clearance and the formation rate of the active drug) and the active drug (to measure its own intrinsic clearance). Use specific enzyme inhibitors to identify contributing isoforms.
- Account for Instability: Include controls to correct for non-enzymatic degradation in buffer. The key output is the formation clearance (
CLint,form) of the active drug from the prodrug and the elimination clearance (CLint,elim) of the active drug.
Table 1: Key In Vitro Parameters for Prodrug PBPK Model Input
| Parameter | Symbol | Typical Experiment | Common Issue & Fix |
|---|---|---|---|
| Prodrug Systemic Clearance | CLint,prodrug |
Incubation in HLM/Hepatocytes | Non-specific binding correction often overlooked. Use measured fumic. |
| Active Drug Formation Clearance | CLint,form |
HLM/HIM incubation measuring active drug appearance. | Ensure assay quantifies both prodrug loss and metabolite formation. |
| Active Drug Elimination Clearance | CLint,elim |
Incubation of synthesized active drug in HLM. | May need to be scaled from recombinant enzyme systems if direct measurement is noisy. |
| Fraction Absorbed | Fa |
Caco-2 permeability assay, or in situ perfusion. | For prodrugs, use the prodrug itself, not just the active moiety. |
| Dissolution Rate | kdis |
USP apparatus in biorelevant media. | Use profile fitting (e.g., Weibull function) for complex formulations. |
Experimental Protocol: Determining Prodrug Activation Kinetics in Human Intestinal S9 Fractions
Objective: To obtain CLint,form and CLint,elim for gut wall metabolism parameterization in a PBPK model.
Materials:
- Test prodrug and authentic standard of active drug.
- Human intestinal S9 fraction (pooled).
- Co-factors: NADPH regenerating system, UDPGA for phase II.
- LC-MS/MS system for quantification.
- Reaction buffer (e.g., phosphate buffer, pH 7.4).
Method:
- Preparation: Thaw S9 fraction on ice. Prepare co-factor solutions and working solutions of test compounds.
- Formation Reaction: In pre-warmed tubes (37°C), mix S9 protein (0.2-0.5 mg/mL), co-factors, and buffer. Initiate reaction by adding prodrug (at least 5 concentrations below estimated Km). Aliquot at multiple time points (e.g., 0, 5, 10, 20, 30 min) into quenching solution (acetonitrile with internal standard).
- Elimination Reaction: Repeat step 2, but initiate by adding the active drug to measure its direct clearance.
- Analysis: Centrifuge quenched samples. Analyze supernatant by LC-MS/MS to quantify prodrug depletion and active drug formation (for step 2) or active drug depletion (for step 3).
- Data Analysis: Plot formation/elimination rate vs. substrate concentration. Fit data to the Michaelis-Menten equation to derive
VmaxandKm. CalculateCLintasVmax/Km. Scale to whole intestine using appropriate scaling factors (S9 protein per gram intestine, intestinal mass).
Research Reagent Solutions Toolkit
Table 2: Essential Materials for Prodrug/Delivery System PBPK Research
| Item | Function in Research |
|---|---|
| Biorelevant Dissolution Media (FaSSGF, FaSSIF-V2, FeSSIF-V2) | Simulates gastric and intestinal fluids for predictive in vitro dissolution testing of formulations. |
| Pooled Human Liver Microsomes (HLM) & Hepatocytes | Gold standard for measuring hepatic metabolic clearance (CLint) and identifying involved CYP enzymes. |
| Pooled Human Intestinal Microsomes (HIM) or S9 | Critical for quantifying gut-wall first-pass metabolism, a key parameter for prodrugs and oral delivery. |
| Transfected Cell Systems (e.g., MDCK-II overexpressing P-gp, BCRP) | To determine transporter kinetics (Km, Jmax) for API or prodrug, informing gut and liver disposition modules. |
| Caco-2 Cell Line | Standard model for assessing passive and active intestinal permeability (Papp), informing the absorption (Fa) parameter. |
| LC-MS/MS System | Essential for sensitive, specific quantification of prodrug and active drug concentrations in complex in vitro and in vivo matrices. |
| PBPK Software Platform (e.g., GastroPlus, Simcyp Simulator, PK-Sim) | Enables integration of in vitro data into a mechanistic physiological framework to simulate and predict in vivo PK. |
Visualizations
Diagram 1: PBPK Modeling Workflow for Prodrug First-Pass Prediction
Diagram 2: Key Processes in Gut Lumen & Enterocyte for Prodrugs
Overcoming Common Pitfalls in PBPK Predictions of First-Pass Metabolism
FAQs & Troubleshooting for PBPK Predictions of First-Pass Metabolism
Q1: My PBPK model consistently underpredicts oral bioavailability (F) compared to clinical data. What are the primary sources of error? A: Underprediction of F often stems from an incomplete representation of first-pass metabolism. Key sources of error include:
- Inaccurate Gut Wall Metabolism: Underestimating CYP3A4/2C9 expression or activity in enterocytes.
- Hepatic Uptake Oversimplification: Assuming passive diffusion only, neglecting active hepatic uptake transporters (e.g., OATP1B1/1B3) that increase clearance.
- Unaccounted Variability: Not incorporating known genetic polymorphisms (e.g., CYP2D6 poor metabolizer prevalence) or demographic factors (age, disease state) into the population simulation.
- Incorrect System Parameters: Using default human physiological values (organ weights, blood flows, enzyme abundances) that do not match your target population.
Q2: During sensitivity analysis, which parameters should I prioritize for variability analysis to understand population outcomes? A: Prioritize parameters with high sensitivity indices (e.g., from Morris or Sobol methods) AND high natural physiological variability. See Table 1.
Table 1: High-Priority Parameters for Variability Analysis in First-Pass PBPK
| Parameter | Physiological Process | Typical Variability (CV%) | Rationale for Prioritization |
|---|---|---|---|
| Hepatic CYP3A4 Abundance | Hepatic Metabolism | 30-50% | High abundance, high variability, key for many drugs. |
| Intestinal CYP3A4 Abundance | Gut Metabolism | >100% | Extreme inter-individual and regional variability in gut. |
| Hepatic Blood Flow (Qh) | Organ Perfusion | 20-30% | Directly affects extraction ratio. Altered in disease. |
| Bile Flow Rate | Enterohepatic Recirculation | 20-40% | Affects re-absorption and secondary exposure peaks. |
| Plasma Protein Binding (fu) | Drug Distribution | 10-50% | Impacts free fraction available for metabolism/uptake. |
| OATP1B1/1B3 Activity | Hepatic Uptake | High (Polymorphic) | Genetic polymorphisms cause large kinetic differences. |
Q3: How can I experimentally verify and refine key input parameters for intestinal metabolism in my model? A: Utilize a tiered in vitro to in vivo extrapolation (IVIVE) protocol.
Experimental Protocol: Refining Gut Wall Metabolism Parameters Objective: Determine accurate in vitro intrinsic clearance (CLint, gut) for IVIVE. Materials: Caco-2 cells, human intestinal microsomes (HIM), relevant cDNA-expressed CYP enzymes, test compound, LC-MS/MS system. Method:
- Cellular System (Caco-2): Culture Caco-2 cells on transwell inserts to form confluent, differentiated monolayers. Apply test compound apically.
- Measurement: Sample from basolateral compartment over time. Quantify parent drug and major metabolites via LC-MS/MS.
- Enzymatic System (HIM): Incubate test compound with HIM and NADPH cofactor. Use chemical inhibitors (e.g., ketoconazole for CYP3A4) to confirm enzyme involvement.
- Kinetic Analysis: Fit depletion data (Caco-2) or metabolite formation data (HIM) to appropriate models (e.g., well-stirred) to obtain CLint.
- Scaling: Scale CLint using appropriate scalars (mg microsomal protein per gram intestine, intestinal mass, etc.). Incorporate into PBPK software and compare prediction to clinical data. Troubleshooting: If scaled clearance still leads to error, check for non-CYP metabolism, inappropriate scalar values, or diffusion-limited kinetics in the cellular system.
Q4: My variability analysis shows unexpected bimodal distributions in AUC. How should I investigate this? A: Bimodality strongly suggests a polymorphic process. Investigate in this order:
- Identify the Driver: Run a focused sensitivity analysis on all polymorphic enzyme/transporter parameters (CYP2D6, CYP2C19, UGT1A1, OATP1B1).
- Correlate with Genotype: If clinical data is available, stratify simulated population by genotype (e.g., PM vs. EM for CYP2D6) and compare to stratified clinical AUCs.
- Refine Population Model: Ensure your virtual population generator correctly represents the allele frequency and phenotype of the polymorphic protein in your target ethnicity.
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in First-Pass Metabolism Research |
|---|---|
| Human Intestinal Microsomes (HIM) | Pooled or individual donor systems to study phase I metabolism specific to the intestinal wall. |
| Transfected Cell Systems (e.g., OATP-HEK293) | To isolate and quantify the kinetics of specific hepatic uptake transporters. |
| Recombinant CYP Enzymes | To determine the enzyme-specific CLint and kinetic constants (Km, Vmax) for a new chemical entity. |
| Specific Chemical Inhibitors (e.g., Ketoconazole, Rifampicin) | To confirm the contribution of specific enzymes (CYP3A4) or transporters (OATP) in in vitro systems. |
| PBPK Software (e.g., GastroPlus, Simcyp, PK-Sim) | Platforms to integrate in vitro data, perform sensitivity/ variability analysis, and simulate population pharmacokinetics. |
| Physiological Database (e.g., ICRP, PHYSPROP) | Sources for accurate, population-specific physiological parameters (organ sizes, blood flows, enzyme abundances). |
Diagram: Workflow for Identifying Error Sources in PBPK Models
Diagram Title: PBPK Model Refinement Workflow
Diagram: Major Pathways in Hepatic First-Pass Metabolism
Diagram Title: Key Hepatic Clearance Pathways
Welcome to the Technical Support Center. This resource provides troubleshooting guidance and FAQs for researchers dealing with missing in vitro parameters, specifically within the context of developing and refining Physiologically-Based Pharmacokinetic (PBPK) models to predict first-pass metabolism.
Troubleshooting Guides & FAQs
Q1: Our laboratory lacks the resources to experimentally determine the intrinsic clearance (CLint) for a new chemical entity. What are the primary in silico strategies to fill this gap for enterocyte metabolism in a "predict-first" PBPK framework?
A: When experimental CLint is unavailable, a tiered in silico approach is recommended.
- Ligand-Based Prediction: Use quantitative structure-activity relationship (QSAR) models. Tools like STARDrop or ADMET Predictor utilize curated datasets of metabolic clearance to predict human hepatic CLint based on molecular descriptors.
- Structure-Based Prediction: If the metabolizing enzyme (e.g., CYP3A4) is known, molecular docking simulations can provide relative binding scores. While not directly quantitative, these scores can be used to rank compounds and infer relative clearance.
- Read-Across: Use data from a close structural analog with known CLint, applying a correction factor based on calculated logP or molecular weight differences. Critical Consideration: Always apply a quantitative uncertainty factor (e.g., 5-10 fold) to these predictions and conduct sensitivity analysis in your PBPK model to understand the impact on the predicted first-pass extraction ratio.
Q2: During the development of a gut wall metabolism PBPK model, we are missing the fraction unbound in the enterocyte (fugut). How can we estimate this parameter, and what is the experimental protocol to validate it?
A: fugut is often assumed to equal hepatic fraction unbound (fuhep) due to similar intracellular protein content, but this can introduce error. A more robust strategy is: Estimation: Use a mechanistic phospholipid binding model based on the compound's lipophilicity (logD at pH 7.4) and pKa, as implemented in tools like Simcyp's "Method 2" for fugut. Validation Protocol:
- Homogenate Preparation: Isolate enterocytes from intestinal slices (human or preclinical species) via EDTA chelation. Homogenize cells in a pH 7.4 buffer (e.g., 100 mM phosphate).
- Equilibrium Dialysis: Use a 96-well equilibrium dialyzer. Load donor side with enterocyte homogenate spiked with test compound. Load receiver side with buffer.
- Incubation & Analysis: Incubate for 4-6 hours at 37°C to reach equilibrium. Quantify compound concentration in both chambers using LC-MS/MS.
- Calculation: fugut = [Concentration]receiver / [Concentration]donor (after equilibrium).
Q3: For modeling first-pass hydrolysis, we need the intestinal luminal degradation rate constant (kdeg). How can we design an experiment to obtain this data if it is missing?
A: kdeg can be determined via an in vitro luminal stability assay. Experimental Protocol:
- Simulated Intestinal Fluid (SIF) Preparation: Prepare FaSSIF-V2 (fasted state simulated intestinal fluid) or FeSSIF-V2 (fed state) as per manufacturer's protocol to mimic human luminal conditions.
- Incubation Setup: In a shaking water bath (37°C, 50 rpm), incubate the test compound in SIF. Use a physiologically relevant pH (e.g., 6.5 for proximal jejunum).
- Sampling: Withdraw aliquots at multiple time points (e.g., 0, 5, 15, 30, 60, 120 min).
- Quenching & Analysis: Immediately quench samples with an equal volume of acetonitrile containing internal standard. Centrifuge and analyze supernatant via LC-MS/MS to determine parent compound remaining.
- Data Fitting: Fit the natural log of percentage remaining vs. time data to a first-order decay model: ln(C) = ln(C0) - kdeg * t.
Q4: How do we handle missing data on inter-individual variability (IIV) for enzyme abundance in the gut for a specific population?
A: When population-specific proteomic data is unavailable:
- Default to Healthy Volunteer Variability: Use the coefficient of variation (CV%) for enzyme abundance (e.g., CYP3A4, UGT1A1) from robust healthy volunteer intestinal biopsy studies. These are often embedded in commercial PBPK platforms.
- Leverage Hepatic Scaling Relationships: For some enzymes, hepatic abundance IIV correlates with intestinal IIV. Apply a scaling factor derived from literature if available.
- Sensitivity & Virtual Population: Conduct a global sensitivity analysis to identify if model AUC or Cmax is highly sensitive to this IIV. If not, the default variability may suffice. If it is critical, generate a virtual population spanning a wide range (e.g., 5th to 95th percentile of known general variability) to bound your predictions.
Table 1: Common In Silico Tools for Predicting Missing Metabolic Parameters
| Tool/Software | Primary Use | Typical Output | Key Limitation |
|---|---|---|---|
| STARDrop | QSAR for CLint, metabolite ID | Predicted human hepatic CLint (µL/min/mg) | Accuracy diminishes for novel scaffolds |
| Simcyp ADME | QSAR & mechanistic modeling | CLint, fu, tissue affinity | Requires compound parameter input |
| GLORY | Site of Metabolism prediction | Likely metabolizing enzyme & site | No quantitative rate prediction |
| MetaTrans | Pathway analysis for biologics | Degradation susceptibility | Focused on peptides/proteins |
Table 2: Key Experimental Assays to Determine Missing Gut-Specific Parameters
| Missing Parameter | Recommended Assay | System Used | Key Output |
|---|---|---|---|
| Enterocytic fu (fugut) | Equilibrium Dialysis | Enterocyte homogenate | Fraction unbound (unitless) |
| Luminal Degradation (kdeg) | Chemical Stability in SIF | FaSSIF/FeSSIF | First-order rate constant (min⁻¹) |
| Transporter Km/Jmax | Uptake in suspended cells | Fresh or cryopreserved human enterocytes | Michaelis-Menten constants |
| Gut Wall CLint | Incubation with intestinal microsomes or S9 fraction | Human intestinal microsomes/S9 | µL/min/mg protein |
Experimental Protocols
Protocol 1: Determining Intrinsic Clearance (CLint) Using Human Intestinal Microsomes Objective: To obtain the metabolic clearance rate of a compound by enterocytic enzymes. Materials: Human intestinal microsomes (HIM), NADPH regeneration system, test compound, LC-MS/MS system. Procedure:
- Prepare incubation mix: 0.1 mg/mL HIM, 1 µM test compound in 100 mM phosphate buffer (pH 7.4).
- Pre-incubate at 37°C for 5 min. Initiate reaction by adding NADPH.
- Aliquot samples at T = 0, 5, 10, 20, 30 minutes into pre-chilled acetonitrile to stop the reaction.
- Centrifuge samples, analyze supernatant via LC-MS/MS to quantify parent compound depletion.
- Fit depletion curve to first-order kinetics: CLint (µL/min/mg) = (k * V) / [Protein], where k is the slope, V is incubation volume.
Protocol 2: Measuring Effective Permeability (Peff) Using Caco-2 Cells Objective: To estimate human jejunal permeability for PBPK model input when in vivo data is absent. Materials: Caco-2 cells (21-25 days post-seeding), transport buffer (HBSS-HEPES), LC-MS/MS. Procedure:
- Wash cell monolayers in 12-well transwell plates with pre-warmed buffer.
- Add donor solution (e.g., apical for A-to-B transport) containing test compound. Receiver chamber contains buffer.
- Incubate on orbital shaker (37°C). Sample from receiver chamber at multiple times (e.g., 30, 60, 90, 120 min), replacing volume.
- Analyze samples. Calculate Papp: Papp (cm/s) = (dQ/dt) / (A * C0), where dQ/dt is transport rate, A is membrane area, C0 is initial donor concentration.
- Apply a correlation equation (e.g., from literature) to scale Caco-2 Papp to human jejunal Peff.
Diagrams
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Context of Filling Data Gaps |
|---|---|
| Cryopreserved Human Enterocytes | Provide intact cellular system for determining uptake CLint, fugut, and transporter kinetics without needing fresh tissue. |
| Human Intestinal Microsomes (HIM) / S9 | Subcellular fractions containing phase I/II enzymes for high-throughput determination of metabolic CLint specific to the gut wall. |
| FaSSIF/FeSSIF Powder | For preparing biorelevant simulated intestinal fluids to assess luminal solubility and chemical stability (kdeg). |
| 96-Well Equilibrium Dialyzer | Enables high-throughput measurement of fraction unbound (fu) in enterocyte homogenate or other matrices. |
| LC-MS/MS System with UHPLC | Essential for sensitive and specific quantification of parent drug depletion in metabolic stability assays and metabolite identification. |
| Validated PBPK Software (e.g., Simcyp, GastroPlus) | Platforms contain built-in systems data (enzyme abundances, IIV, physiology) and tools to incorporate new in vitro data or in silico predictions. |
| QSAR/ADMET Prediction Software License | Provides critical in silico estimates (CLint, permeability) when experimental data is completely missing for initial modeling. |
Technical Support Center: Troubleshooting PBPK Model Implementation
Frequently Asked Questions (FAQs)
Q1: My model consistently over-predicts the plasma concentration of a drug known to undergo extensive first-pass metabolism. Which parameters should I prioritize for refinement? A1: Prioritize refining the tissue-to-plasma partition coefficients (Kp) for the liver and gut, and the intestinal and hepatic transit times. Over-prediction often stems from underestimating the extraction due to inaccurate partitioning (which determines the available drug for metabolism) or overly rapid transit through metabolizing organs.
Q2: During model validation, the predicted vs. observed AUC ratio is outside the acceptable 2-fold range for oral dosing only. What does this indicate? A2: This typically indicates an error in the characterization of first-pass extraction. Focus your troubleshooting on the portal vein absorption model (including enterocyte metabolism) and the hepatic clearance model. Verify the in vitro-in vivo extrapolation (IVIVE) of intrinsic clearance and the effective permeability used for the gut.
Q3: How sensitive are first-pass metabolism predictions to the method used for estimating tissue partition coefficients (Kp)? A3: Highly sensitive. The choice between mechanistic (e.g., Poulin & Theil, Berezhkovskiy) and empirical methods can lead to significant variance in predicted hepatic and gut tissue concentrations, directly impacting the metabolic rate. Use the table below for comparison.
Table 1: Common Methods for Estimating Tissue:Plasma Partition Coefficients (Kp)
| Method | Key Principle | Best For | Limitation for First-Pass |
|---|---|---|---|
| Rodgers & Rowland | Mechanistic; based on tissue composition, drug lipophilicity (logP) and pKa. | Neutral, zwitterionic, and bases. | May require scaling factors for highly bound drugs in liver. |
| Poulin & Theil | Mechanistic; extends Rodgers & Rowland with different formalism. | Neutral and acidic compounds. | Predictions for adipose tissue can be problematic. |
| Berezhkovskiy | Conserves mass balance for perfused tissue models. | Integration into perfusion-limited organ models. | Requires accurate plasma and red blood cell binding data. |
| In Vitro-In Vivo Extrapolation (IVIVE) | Uses measured tissue slice or homogenate data. | Compounds with atypical distribution. | Experimentally intensive, scaling factors needed. |
Q4: What are the critical experimental protocols for validating intestinal transit and metabolism parameters? A4:
- In Situ Single-Pass Intestinal Perfusion (SPIP): Determines effective permeability (Peff) and assesses intestinal metabolism. Cannulate a segment of rat jejunum, perfuse with drug solution, and measure disappearance from lumen and appearance in mesenteric blood.
- In Vitro Caco-2 Permeability Assay: Correlates with human Peff. Culture human colorectal adenocarcinoma cells on transwell inserts, measure apical-to-basolateral transport over time.
- Hepatocyte & Microsome Incubations: For intrinsic clearance (CLint). Incubate drug with liver microsomes or suspended hepatocytes, measure substrate depletion over time to calculate in vitro CLint, followed by IVIVE using liver size and microsomal protein per gram of liver scaling factors.
Q5: The model fails to capture dual-peak pharmacokinetic profiles after oral administration. Which transit time model should I re-evaluate? A5: Re-evaluate the gastric emptying and intestinal transit model. A simple first-order transit may be insufficient. Implement a more physiologically based model like the Compartmental Transit Time (CTT) model or a delayed-absorption model (e.g., a series of compartments) to account for complex emptying patterns.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Key PBPK Refinement Experiments
| Item | Function | Example / Specification |
|---|---|---|
| Caco-2 Cell Line | In vitro model of human intestinal permeability and efflux transport. | ATCC HTB-37, passage numbers 25-45 for optimal differentiation. |
| Cryopreserved Hepatocytes | Gold standard for in vitro measurement of hepatic metabolic intrinsic clearance (CLint). | Human, 3-donor pooled, high viability (>80%), gender-specified. |
| Liver Microsomes | Subcellular fraction containing CYP450 enzymes for metabolic stability assays. | Human, 50-donor pooled, supplemented with NADPH-regenerating system. |
| Dulbecco's Modified Eagle Medium (DMEM) | Cell culture medium for maintaining Caco-2 cells. | High glucose, with L-glutamine, without sodium pyruvate. |
| Transwell Permeable Supports | Polycarbonate membrane inserts for culturing polarized cell monolayers for transport assays. | 12-well plate, 1.12 cm² surface area, 3.0 µm pore size. |
| Simcyp Simulator or GastroPlus | Leading PBPK software platforms for integrating in vitro data, refining models, and simulating first-pass metabolism. | Academic licenses available; include validated compound, demographic, and enzyme databases. |
Experimental Workflow for Model Refinement
First-Pass Metabolism Pathway in PBPK Context
Technical Support Center: Troubleshooting PBPK Modeling for First-Pass Metabolism
FAQs & Troubleshooting Guides
Q1: My PBPK model under-predicts the observed AUC increase for my BCS Class II drug when administered with a high-fat meal. What are the key model parameters to investigate? A: This typically indicates an incomplete characterization of food's physiological effects on first-pass metabolism. Focus on these parameters:
- Gastric Emptying Rate: A high-fat meal slows gastric emptying, altering drug arrival time to the intestine and liver. Ensure your model uses fed-state parameters.
- Hepatoportal Blood Flow: Food increases splanchnic blood flow. Verify your model implements a postprandial increase in portal vein blood flow (~30-50% rise).
- Bile Salt Secretion: Fat stimulates bile flow, which is critical for solubilizing lipophilic drugs. Confirm your dissolution model accounts for enhanced solubility due to micellar solubilization.
- Enterocyte-Based Metabolism: For drugs like midazolam, food may directly modulate CYP3A4 activity in the gut wall. Review if your intestinal metabolism model includes potential food-induced inhibition or induction.
Q2: During the simulation of a nonlinear, saturable metabolic pathway (e.g., phenytoin), my model fails to converge at higher doses. How can I resolve this? A: Non-convergence often stems from numerical instability in solving the Michaelis-Menten equations. Follow this protocol:
- Verify Initial Estimates: Ensure your initial estimates for Vmax and Km are physiologically plausible and within the correct order of magnitude.
- Implement a Hybrid Solver: Use a stiff solver (e.g., Rosenbrock) for the metabolic pathway while using a standard solver for the rest of the system.
- Dose Ramping: Instead of simulating a high dose directly, run sequential simulations with incrementally increasing doses to provide a stable solution path.
- Check Mass Balance: Implement an output to track total drug mass (absorbed, metabolized, excreted) at each time step to identify when/where mass is being lost.
Q3: My predicted DDI magnitude for a time-dependent CYP3A4 inhibitor (e.g., erythromycin) is much lower than clinical observations. What is the most likely missing mechanism? A: The discrepancy likely arises from not modeling the mechanism-based inactivation (MBI) process correctly. You must distinguish between reversible inhibition and MBI.
Experimental Protocol for MBI Parameter Estimation (In Vitro to In Vivo):
- In Vitro Incubation: Pre-incubate human liver microsomes with the inhibitor (multiple concentrations) and NADPH for varying times (0-30 min).
- Dilution & Activity Probe: Dilute the mixture greatly (e.g., 20-fold) to minimize reversible inhibition, then add a specific CYP probe substrate (e.g., midazolam for CYP3A4).
- Kinetic Analysis: Plot remaining enzyme activity vs. pre-incubation time. Fit data to derive kinact (maximum inactivation rate constant) and KI (inhibitor concentration producing half-maximal inactivation).
- In Vitro to In Vivo Scaling: Incorporate these parameters into your PBPK model using a kinact/KI ratio. The model must dynamically deactivate the enzyme pool and account for re-synthesis (zero-order synthesis rate, ksynth, with a typical hepatic enzyme half-life of ~24-48 hours).
Key Research Reagent Solutions
| Reagent / Material | Function in PBPK-Focused Research |
|---|---|
| Cryopreserved Human Hepatocytes | Gold standard for determining intrinsic clearance (CLint) and studying induction/ inhibition of metabolism. |
| Human Liver Microsomes (HLM) | Used for high-throughput determination of metabolic stability, reaction phenotyping, and kinetic parameters (Km, Vmax, KI). |
| Transfected Cell Systems (e.g., CYP-Overexpressing Caco-2) | To isolate and study the kinetics of a single metabolic pathway or transporter, reducing complexity. |
| Physiologically Relevant Dissolution Media (FaSSIF/FeSSIF) | Simulates fasted and fed state intestinal fluids to measure realistic dissolution profiles for solubility-limited drugs. |
| Specific CYP & Transporter Probe Substrates/Inhibitors | Essential for reaction phenotyping experiments to identify enzymes/transporters involved in a drug's first-pass metabolism. |
Quantitative Data Summary: Key Parameters for Complex Scenarios
Table 1: Typical Physiological Changes in Fed vs. Fasted State Relevant to PBPK
| Parameter | Fasted State | High-Fat Fed State | Change |
|---|---|---|---|
| Gastric Emptying Half-Time | 10-20 min | 45-90 min | Slowed |
| Portal Vein Blood Flow | ~1.05 L/min | ~1.4 L/min | +30-50% |
| Hepatic Blood Flow | ~1.5 L/min | ~1.5 L/min | Minimal |
| Bile Salt Secretion | Low | High | Increased |
| Intestinal pH (Duodenum) | ~6.5 | ~5.0 | Decreased |
Table 2: Common Nonlinear Kinetics Examples in First-Pass Metabolism
| Drug | Primary Enzyme | Approximate Km (µM) | Clinical Dose Range Where Nonlinearity May Occur |
|---|---|---|---|
| Phenytoin | CYP2C9 | ~5-10 | > 300 mg/day |
| Sildenafil | CYP3A4 | ~3.6 | > 50 mg |
| Ethanol | CYP2E1, ADH | ~100-200 (CYP2E1) | Variable |
| Theophylline | CYP1A2 | ~40 | > 400 mg/day |
Visualizations
Title: PBPK Workflow for Modeling Food Effects on First-Pass Metabolism
Title: Reversible vs. Mechanism-Based Inhibition in DDI Models
Best Practices for Modeling Special Populations (e.g., Hepatic Impairment)
Troubleshooting Guides and FAQs
Q1: My PBPK model under-predicts systemic exposure in subjects with moderate hepatic impairment. What are the primary physiological parameters to verify? A: First, ensure the following parameters are accurately scaled for the impairment Child-Pugh (CP) class: Hepatic blood flow, hematocrit, plasma protein binding (especially albumin), and functional liver volume/cell mass. For CP-B, hepatic blood flow is typically reduced by 20-30%, functional liver mass by 30-50%, and albumin by ~20%. Verify that the reduction in intrinsic clearance (CLint) due to CYP enzyme activity is informed by in vitro or clinical data, not just a simple linear scaling with liver size.
Q2: When modeling a drug with high first-pass metabolism, how should I approach changes in portal vein blood flow and shunt fraction in cirrhosis? A: In cirrhosis, portal hypertension leads to intra- and extra-hepatic shunting, diverting drug away from metabolizing enzymes. You must modify the model structure to include a shunt pathway. The fraction of portal blood shunted directly to systemic circulation can range from 20% (mild cirrhosis) to 60% (severe). This significantly increases bioavailability for high-extraction drugs.
Q3: What is the best practice for integrating in vitro data on CYP inhibition or induction potential from hepatocytes of impaired livers? A: Use hepatocytes isolated from donors with hepatic impairment (available from vendors like BioIVT or Lonza) to derive relative changes in baseline CYP activity and induction/inhibition response compared to healthy hepatocytes. Incorporate these as scaling factors to the healthy CLint values in your model. Do not assume the fractional change is uniform across all CYPs.
Q4: How can I verify my model's performance for a special population if clinical data is very limited? A: Employ a "predict-first" approach. Prior to clinical data, predict the PK profile using your PBPK model built solely on in vitro and healthy volunteer data. Document the prediction and uncertainty. Once sparse data is available, perform a limited calibration only on key system-specific parameters (e.g., shunt fraction), not on drug-specific parameters like CLint. Assess if the prediction falls within the 90% prediction interval.
Key Experiment Protocols
Protocol 1: In Vitro-In Vivo Extrapolation (IVIVE) for Hepatic Impairment CLint Scaling
Objective: To scale intrinsic clearance (CLint) from healthy to hepatic impairment conditions. Methodology:
- Materials: Cryopreserved human hepatocytes from healthy (N≥5) and hepatically impaired (CP-A, B, C; N≥3 each) donors. Test drug at sub-Km concentration.
- Incubation: Perform substrate depletion assays in suspension hepatocytes (0.5 million cells/mL) for 120 minutes.
- Analysis: Determine in vitro CLint (µL/min/million cells) from depletion slope.
- Scaling Factor: Calculate the geometric mean ratio of CLint (Impaired/Healthy) for each CP class.
- Implementation: Apply this ratio as a scalar to the healthy human CLint value in the PBPK model.
Protocol 2: Determining Portal-Systemic Shunt Fraction
Objective: To estimate the fraction of portal blood bypassing the liver sinusoids. Methodology (Pharmacokinetic Method):
- Administer a high-extraction ratio compound (e.g., sorbitol, cholate) intravenously and orally.
- Measure the systemic bioavailability (F).
- Calculate using the well-stirred liver model: F = fa * fg * [1 - E], where E = (CLh / Qh). For a high-extraction drug, fa and fg ~1, so E ≈ 1 - F.
- Derive Shunt: The observed E in cirrhosis will be lower than in health. The shunt fraction (S) can be estimated as: S = (Ehealthy - Ecirrhosis) / E_healthy. This requires data from a separate healthy cohort.
Data Tables
Table 1: Typical Physiological Changes in Chronic Liver Disease (Child-Pugh Classes)
| Physiological Parameter | CP-A (Mild) | CP-B (Moderate) | CP-C (Severe) | Source/Note |
|---|---|---|---|---|
| Hepatic Blood Flow (% of Healthy) | 90-100% | 70-85% | 50-70% | Model input range |
| Functional Liver Cell Mass (%) | 70-80% | 50-70% | 30-50% | Based on imaging & CYP activity |
| Serum Albumin (g/dL) | 3.5 - <4.0 | 2.8 - <3.5 | <2.8 | Clinical CP score component |
| Portal-Systemic Shunt Fraction | 5-20% | 20-40% | 40-60% | Pharmacokinetic estimation |
| CYP3A4 Activity (% of Healthy) | 70-80% | 40-60% | 20-40% | In vitro hepatocyte data |
Table 2: Common Modeling Scenarios and Recommended Adjustments
| Drug Property | Primary HI Effect | Key Model Adjustment | Troubleshooting Tip |
|---|---|---|---|
| High Extraction, Low Solubility | Reduced first-pass, shunting | ↓ Liver blood flow, ↑ Shunt fraction | If prediction too low, check shunt and enterocyte metabolism (f_g). |
| Low Extraction, Albumin Bound | ↑ Free fraction, ↓ CLint | ↓ CLint, ↑ Free fraction in plasma | Measure free drug concentration if possible. Binding change may offset CLint change. |
| Renally Eliminated (with Metabolites) | Compromised renal function | ↓ GFR per CP class (e.g., CP-B: -25%) | Account for potential metabolite accumulation. |
Visualizations
Title: PBPK Workflow for Hepatic Impairment
Title: Altered First-Pass Pathways in Liver Cirrhosis
The Scientist's Toolkit: Research Reagent Solutions
| Item/Vendor | Function in HI Modeling | Notes |
|---|---|---|
| Cryopreserved Hepatocytes (Impaired Donors) (e.g., BioIVT, Lonza) | Provide direct in vitro data on CYP activity, CLint, and induction response in HI. | Request donors with Child-Pugh classification and full medical history. |
| Human Liver Microsomes (HLM) from HI Donors (e.g., XenoTech) | Assess metabolic stability and reaction phenotyping in HI. | Useful but lacks full cellular transport and regulatory context. |
| Plasma from HI Donors (e.g., Tennessee Blood Bank) | Determine disease-specific plasma protein binding (f_u). | Critical for highly bound drugs; albumin and AAG levels vary. |
| Specialized PBPK Software (e.g., GastroPlus, Simcyp, PK-Sim) | Contain pre-built HI population libraries with scaled physiology. | Verify the underlying assumptions of the library before use. |
| Physiologically-based PD (PBPD) Models (e.g., DILIsym) | Extend PBPK to predict pharmacodynamic or toxicity outcomes in HI. | Essential for drugs with narrow therapeutic index. |
Validating PBPK Predictions: Standards, Comparisons, and Regulatory Acceptance
FAQs & Troubleshooting Guide
Q1: During PBPK model validation for first-pass metabolism prediction, my observed plasma concentration data falls outside the 95% confidence interval of the simulated data. What are the key acceptance criteria I should check first?
A: First, systematically assess the following standard acceptance criteria benchmarks. The table below summarizes quantitative thresholds for key metrics.
Table 1: Key Acceptance Criteria for PBPK Model Validation (First-Pass Focus)
| Metric | Formula / Description | Acceptance Threshold | Diagnostic Action if Failed |
|---|---|---|---|
| Predicted/Observed (P/O) Ratio | Ratio of Predicted to Observed AUC or Cmax | 0.8 - 1.25 (1.5 for enzymes with high variability) | Check enzyme abundance (ISEF), fraction unbound (fu), or intestinal permeability inputs. |
| Average Fold Error (AFE) | 10^(Σ log(P/O) / n) | 0.7 - 1.43 | Bias towards over/under-prediction. Calibrate systemic clearance or fm. |
| Absolute Average Fold Error (AAFE) | 10^(Σ |log(P/O)| / n) | < 2.0 | Overall accuracy error. Re-evaluate intrinsic clearance (CLint) values from in vitro data. |
| Visual Predictive Check (VPC) | % of observed points within 90% or 95% prediction interval | >90% of data points within the PI | Model misspecification. Consider inter-individual variability (IIV) on first-pass processes (e.g., CYP abundance, gastric emptying). |
| Mean Squared Prediction Error (MSPE) | Σ (Predicted - Observed)² / n | Context-dependent; compare to a "naive" model. | High error suggests poor predictive performance. Review the relevance of in vitro-in vivo extrapolation (IVIVE) assumptions. |
Q2: I am using in vitro hepatocyte data to inform hepatic clearance in my PBPK model, but the model consistently under-predicts first-pass extraction (over-predicts oral AUC). What are the primary troubleshooting steps?
A: This common issue often relates to the scaling of in vitro data. Follow this experimental protocol for systematic troubleshooting.
Experimental Protocol: Refining Hepatic CLint from In Vitro Data
- Re-assess Binding Corrections: Confirm experimental fraction unbound in microsomes/incubation (fuinc) was measured and applied correctly. Use the revised well-stirred model: CLintivive = CLintobs / fuinc.
- Verify Scaling Factors: Ensure appropriate scaling factors (e.g., microsomal protein per gram of liver (MPPGL), hepatocellularity (HPGL), and liver weight) are current and population-specific.
- Check for Inter-System Extrapolation Factors (ISEF): Apply enzyme-specific ISEFs to calibrate recombinant enzyme system data to native tissue activity. Omit if using human hepatocytes.
- Evaluate Non-Standard Metabolism: Investigate contributions from non-CYP enzymes (e.g., UGTs, esterases) or extrahepatic metabolism (e.g., gut wall) not captured in hepatocyte assays.
- Protocol Detail: For step 1, the in vitro CLint determination should use substrate depletion or metabolite formation assays with at least 6 concentrations (covering Km range) and triplicate measurements. Incubation times must be within linear range for metabolite formation/deplation.
Q3: How do I define acceptance criteria for a PBPK model predicting first-pass metabolism when clinical data is very limited (e.g., only single-dose PK)?
A: With limited data, employ a tiered, weight-of-evidence approach beyond simple P/O ratios.
- Leverage IVIVE Consistency: The model should reasonably predict in vivo hepatic clearance from in vitro data (AFE < 2) before introducing complex first-pass mechanisms.
- Sensitivity Analysis: Perform a global sensitivity analysis (e.g., Sobol method) to confirm that first-pass parameters (e.g., gut permeability, intestinal CYP3A4 abundance) are influential for the oral AUC prediction.
- Virtual Population (VPop) Check: Ensure the simulated VPop's range of PK parameters (AUC, Cmax, Tmax) encompasses the single-dose observed data.
- Qualitative Trend Validation: The model must correctly predict the directional change in exposure when co-administered with a known enzyme inhibitor or inducer (if such in vivo data exists).
Research Reagent Solutions Toolkit
Table 2: Essential Reagents & Materials for PBPK-Focused First-Pass Metabolism Research
| Item | Function / Application | Key Consideration |
|---|---|---|
| Cryopreserved Human Hepatocytes | Gold standard for determining intrinsic clearance (CLint) and metabolic stability. | Pooled donors recommended to capture average enzyme activity. Check viability (>80%) post-thaw. |
| Human Liver Microsomes (HLM) | Contains major CYP enzymes for reaction phenotyping and CLint determination. | Use pooled HLM from a sufficient donor pool (e.g., ≥50) for general predictions. |
| Recombinant CYP Enzymes (rCYP) | Used for reaction phenotyping to identify enzymes responsible for metabolism. | Requires Inter-System Extrapolation Factor (ISEF) for quantitative scaling. |
| Caco-2 Cell Line | Standard in vitro model for predicting human intestinal permeability (Peff). | Passage number and culture conditions significantly impact transporter expression. |
| Specific Chemical Inhibitors (e.g., Ketoconazole for CYP3A4) | Used in reaction phenotyping studies to quantify fraction metabolized (fm) by specific pathways. | Verify inhibitor specificity and concentration to avoid off-target effects. |
| LC-MS/MS System | Essential for quantifying drug and metabolite concentrations in in vitro assays and biological samples. | Method must be validated for sensitivity (LLOQ) and specificity in complex matrices. |
| PBPK Software (e.g., GastroPlus, Simcyp, PK-Sim) | Platform for integrating in vitro data, system parameters, and performing simulations. | Choice influences available system databases and customization options for gut physiology. |
Key Methodologies & Visualizations
Workflow for PBPK Model Validation of First-Pass Metabolism
Key Pathways Influencing First-Pass Metabolism in PBPK
Technical Support & Troubleshooting Center
FAQ: General Concepts
Q1: In the context of my thesis on first-pass metabolism prediction, what is the fundamental difference between the two approaches? A: Traditional Allometric Scaling (AS) is an empirical, top-down method that extrapolates pharmacokinetic parameters (like clearance) from animals to humans using a power-law equation based on body weight. It assumes anatomical/physiological similarity across species. Physiologically-Based Pharmacokinetic (PBPK) modeling is a mechanistic, bottom-up approach. It integrates system-specific (human physiology), drug-specific (chemical properties), and enzyme/transporter kinetics data to simulate drug concentration-time profiles in specific organs, including the gut wall and liver, which govern first-pass extraction.
Q2: When validating my first-pass PBPK model, the predicted hepatic extraction ratio is consistently overestimated. What are the primary troubleshooting steps? A: Follow this systematic guide:
- Verify Input Parameters: Re-check the intrinsic clearance (CLint) values from in vitro assays (e.g., human liver microsomes). Ensure correct scaling factors (microsomal protein per gram of liver, hepatocellularity) are applied.
- Assess Binding Corrections: Confirm the accuracy of fraction unbound in blood (fu) and microsomal incubations (fu_inc). Underestimating binding leads to overprediction of CLint.
- Review Model Physiology: Ensure liver blood flow (Qh) and portal vein flow values are appropriate for your simulated population (e.g., healthy vs. cirrhotic).
- Consider Transport: For substrates of hepatic uptake transporters (e.g., OATP1B1), the "well-stirred" liver model may be insufficient. Incorporate active uptake kinetics or switch to a "parallel tube" or "dispersion" model.
- Check for Inhibition: Ensure no unintended inhibition is occurring in your in vitro CLint assay due to high drug concentrations.
Q3: My allometric scaling predictions for human oral bioavailability (F) fail when the compound undergoes significant intestinal metabolism. How can I address this? A: Traditional AS based on intravenous data alone cannot predict first-pass loss in the gut. To troubleshoot:
- Integrate Data: Incorporate preclinical oral bioavailability data from multiple species. Plot the fraction absorbed (Fa) and the gut wall extraction (Eg) against species-specific physiological factors (e.g., intestinal CYP450 abundance).
- Use a Hybrid Approach: Apply AS to predict systemic clearance (CL) and volume of distribution (Vd). Then, use a minimal PBPK model (e.g., a gut compartment with enterocyte metabolism) to separately estimate intestinal first-pass extraction, informed by in vitro Caco-2 or intestinal microsome data.
Experimental Protocol: Key Methodologies
Protocol 1: Determining In Vitro Intrinsic Clearance (CLint) for Hepatic Input Objective: To obtain the primary kinetic parameter for scaling to hepatic metabolic clearance.
- Incubation: Prepare human liver microsomes (HLM, 0.5 mg/ml protein) in phosphate buffer (pH 7.4). Add test compound (1 µM, range for Km determination) and pre-incubate for 5 min at 37°C.
- Reaction Initiation: Start reaction by adding NADPH (1 mM final concentration). Run duplicate samples.
- Termination: At predetermined timepoints (e.g., 0, 5, 10, 20, 30 min), remove aliquots and quench with ice-cold acetonitrile containing internal standard.
- Analysis: Centrifuge, analyze supernatant via LC-MS/MS to determine parent compound depletion.
- Calculation: Fit depletion curve to first-order decay model: CLint = (ln(C0/Ct) / (t * [microsomal protein])).
Protocol 2: Performing Allometric Scaling for Human IV Clearance Prediction Objective: To extrapolate total body clearance from preclinical species to humans.
- Data Collection: Obtain steady-state volume of distribution (Vss) and clearance (CL) from intravenous studies in at least three animal species (e.g., rat, dog, monkey).
- Plotting: On log-log paper or software, plot CL vs. body weight (BW) for each species.
- Fitting: Apply the allometric equation: CL = a * BW^b. Perform linear regression on the log-transformed data: log(CL) = log(a) + b * log(BW).
- Prediction: For a standard 70 kg human, calculate predicted CLhuman = a * (70)^b.
- Correction (Optional): Apply species-invariant time methods (e.g., Brain Weight, Maximum Life Span Potential) as needed.
Data Presentation
Table 1: Quantitative Comparison of PBPK vs. Allometric Scaling for First-Pass Prediction
| Feature | Traditional Allometric Scaling | PBPK Modeling |
|---|---|---|
| Core Approach | Empirical, top-down extrapolation. | Mechanistic, bottom-up simulation. |
| Primary Data Input | In vivo PK parameters from animals. | In vitro drug properties, in silico parameters, system physiology. |
| Prediction of First-Pass | Cannot directly predict. Requires separate in vivo oral data for scaling. | Directly simulates gut wall and hepatic extraction via organ models. |
| Species Extrapolation Basis | Body weight (allometric exponent). | Species-specific physiological parameters (organ weights, blood flows, enzyme abundances). |
| Ability to Probe Mechanisms | None. "Black-box" output. | High. Can isolate contribution of specific enzymes, transporters, or physiological changes. |
| Typical Prediction Accuracy (for Hepatic CL) | Often within 2-fold error for passively cleared drugs. | Can achieve <1.5-fold error with robust in vitro-in vivo extrapolation (IVIVE). |
| Handling of Drug-Drug Interactions (DDI) | Cannot predict. | Excellent for predicting enzyme/transporter-mediated DDIs at first-pass organs. |
| Requirement for Animal Data | Mandatory (multiple species). | Minimal to none for a "first-in-human" prediction. |
Table 2: Research Reagent Solutions Toolkit
| Item | Function in First-Pass Metabolism Research |
|---|---|
| Human Liver Microsomes (HLM) | Pooled subcellular fraction containing CYP450s and other enzymes; used to determine hepatic CLint. |
| Human Intestinal Microsomes (HIM) | Used to determine metabolic CLint specific to the gut wall (e.g., for CYP3A4 substrates). |
| Recombinant CYP450 Enzymes | Expressed single enzymes (e.g., rCYP3A4) to identify specific isoforms responsible for metabolism. |
| Caco-2 Cell Line | Model of human intestinal epithelium; used to study permeability and potential transporter effects. |
| Hepatocytes (Cryopreserved) | Gold-standard in vitro system with full complement of enzymes and transporters; used for CLint and uptake studies. |
| NADPH Regenerating System | Provides essential cofactor for CYP450-mediated oxidative metabolism reactions in vitro. |
| Specific Chemical Inhibitors (e.g., Ketoconazole for CYP3A4) | Used in in vitro incubations to phenotype the enzymes involved in metabolite formation. |
Mandatory Visualizations
Title: PBPK Model First-Pass Metabolism Pathway
Title: PBPK vs Allometric Scaling Workflow Comparison
FAQs & Troubleshooting
Q1: What are the minimum validation criteria for a PBPK model to be submitted to the FDA or EMA for bioavailability (BA) or bioequivalence (BE) waiver requests?
A: Both agencies require robust model validation. Key criteria include:
- Prior Knowledge: The model must be built and verified using established physiology, pharmacokinetic principles, and credible drug-specific data (e.g., in vitro dissolution, permeability, metabolic stability).
- External Validation: The model must demonstrate predictive performance by comparing simulations against observed clinical pharmacokinetic data not used in model development (e.g., from a different study population, dosage form, or dose level).
- Sensitivity Analysis: A formal sensitivity analysis must identify critical model parameters (e.g., solubility, particle size, CYP enzyme affinity) that significantly impact the predicted BA. This defines the model's applicability domain.
- Acceptance Criteria: Predictions for key exposure metrics (AUC, Cmax) should generally fall within a pre-defined threshold (e.g., within 1.25-fold or 2-fold) of observed data, depending on the application's risk.
Q2: My PBPK model accurately predicts AUC but consistently under-predicts Cmax for an IR formulation. What could be the issue?
A: This is a common troubleshooting scenario. The issue likely lies in the dissolution or absorption model setup.
- Check 1: In Vitro Dissolution Data & Method. Ensure the in vitro dissolution method is biorelevant and the data is accurately represented in the model (e.g., using a suitable function like Weibull or Johnson). An overly slow dissolution profile will under-predict Cmax.
- Check 2: Gastric Emptying & Intestinal Transit. Review the physiological parameters for gastric emptying (especially for immediate-release solids). The default transit models may be too slow. Consider sensitivity analyses on these parameters.
- Check 3: Precipitation & Supersaturation. For weakly basic drugs, if supersaturation is not modeled or precipitation is over-predicted, Cmax will be under-predicted. Review the precipitation time and rate constants.
Q3: For a BCS Class II drug, how do EMA and FDA differ in their expectations for using PBPK to justify a biowaiver for a post-approval change (e.g., particle size distribution)?
A: While both encourage the use, there are nuanced differences in emphasis.
| Agency | Key Perspective & Requirements |
|---|---|
| FDA | Stronger emphasis on the integration of in vitro dissolution data into the PBPK model. The model must demonstrate that dissolution is the rate-limiting step for absorption. A virtual bioequivalence trial comparing the changed vs. reference product is often required, with predefined equivalence bounds (e.g., 90% CI for AUC and Cmax ratios within 80-125%). |
| EMA | Also accepts PBPK for this purpose. Places significant weight on a thorough sensitivity analysis to establish the "safe space" – the range of critical material attributes (like particle size) and formulation parameters within which bioequivalence is predicted. The justification relies heavily on demonstrating the change remains within this "safe space." |
Q4: When modeling first-pass metabolism for bioavailability prediction, my systemic clearance is correct, but BA is over-predicted. What specific parameters should I investigate?
A: This points to an under-prediction of pre-systemic (first-pass) extraction.
- Investigate 1: Hepatic Uptake. If the drug is a substrate for active hepatic uptake transporters (e.g., OATP1B1/1B3), ensure the in vitro uptake clearance (
CLint,uptake) is correctly scaled and incorporated. Missing this can vastly over-predict BA. - Investigate 2: Gut Metabolism. For drugs metabolized by CYP3A4 or UGTs in the gut, verify the abundance and activity of the enzyme in the enterocyte compartment. Default values may be insufficient for high-extraction compounds.
- Investigate 3: Portal Vein Blood Flow. Confirm the model uses appropriate, physiology-based values for portal vein and hepatic blood flows, as these directly determine extraction ratio.
Experimental Protocols
Protocol 1: In Vitro Assay for Determining Key PBPK Input Parameters
This protocol outlines key experiments to generate data for a PBPK model focused on first-pass metabolism and bioavailability.
Title: Determination of Hepatic Metabolic and Uptake Clearance for PBPK Inputs.
Objective: To obtain in vitro intrinsic clearance (CLint) for metabolism and active hepatic uptake to scale to in vivo organ clearance.
Materials: See "Research Reagent Solutions" table below.
Methodology:
- Microsomal Metabolic Stability: Incubate the drug (1 µM) with human liver microsomes (0.5 mg/mL) in potassium phosphate buffer (pH 7.4) with NADPH regenerating system. Take aliquots at 0, 5, 15, 30, and 60 minutes. Stop reaction with acetonitrile containing internal standard.
- Analysis: Quantify parent drug loss via LC-MS/MS. Calculate
CLint,met(µL/min/mg protein) using the substrate depletion method. - Transfected Cell Uptake Assay: Culture HEK293 cells stably expressing OATP1B1. Incubate cells with the drug (1 µM) in uptake buffer (Hanks' Balanced Salt Solution, HBSS) at 37°C for 2 minutes. Include vector-control cells.
- Termination & Analysis: Rapidly wash cells with ice-cold HBSS. Lyse cells and quantify intracellular drug accumulation via LC-MS/MS. Subtract uptake in control cells to determine OATP-mediated uptake. Calculate
CLint,uptake(µL/min/mg protein). - In Vitro-In Vivo Extrapolation (IVIVE): Scale microsomal
CLint,metto hepatic metabolic clearance using scaling factors (microsomal protein per gram of liver, liver weight). Scale cellularCLint,uptakeusing relevant cellular protein scaling factors.
Protocol 2: Virtual Bioequivalence (VBE) Study for a Formulation Change
Title: PBPK Workflow for Virtual Bioequivalence Assessment.
Objective: To simulate the pharmacokinetics of a new formulation (Test) against the reference formulation to support a biowaiver.
Methodology:
- Base Model Development & Validation: Develop a full PBPK model for the reference formulation using all available in vitro and in vivo data. Validate the model against a clinical PK study not used for model building.
- Parameterization of Test Formulation: Modify the model's dissolution component to reflect the in vitro dissolution profile of the new test formulation (e.g., different particle size distribution or excipient ratio).
- Virtual Population: Simulate a virtual population (
N=100-1000) demographically matching the target patient population (age, weight, sex, genotype if relevant) using the simulator's population builder. - Virtual Trial Execution: Run simulations for both the Reference and Test formulations in the same virtual subjects under the same conditions (fasted/fed state, dose).
- Statistical Comparison: Extract simulated AUC and Cmax for both formulations. Calculate the geometric mean ratio (Test/Reference) and its 90% confidence interval. Apply bioequivalence criteria (typically 80-125%).
- Sensitivity & "Safe Space" Analysis: Conduct a multivariate sensitivity analysis around the critical quality attribute (e.g., particle size) to define the range where bioequivalence is maintained.
Diagrams
PBPK Model Validation & Troubleshooting Workflow
Key Pathways Determining Oral Bioavailability (BA)
Research Reagent Solutions
| Item | Function in PBPK-Related Research |
|---|---|
| Human Liver Microsomes (HLM) | Subcellular fraction containing membrane-bound CYP enzymes. Used in metabolic stability assays to determine intrinsic metabolic clearance (CLint,met). |
| Transporter-Transfected Cell Lines (e.g., HEK293-OATP1B1, MDCKII-Pgp) | Engineered cells overexpressing a single human transporter. Critical for isolating and quantifying transporter-specific uptake or efflux activity for model input. |
| Biorelevant Dissolution Media (FaSSIF, FeSSIF) | Simulated intestinal fluids that mimic the fasting and fed state. Provide physiologically relevant in vitro dissolution profiles for more accurate PBPK absorption modeling. |
| NADPH Regenerating System | Supplies a constant concentration of NADPH, the essential cofactor for CYP-mediated oxidation reactions in microsomal incubations. |
| LC-MS/MS System | Gold-standard analytical platform for quantifying drug concentrations in complex biological matrices (e.g., incubation media, cell lysates, plasma) with high sensitivity and specificity. |
| PBPK Software Platform (e.g., GastroPlus, Simcyp Simulator, PK-Sim) | Integrated software that incorporates physiological, drug, and formulation data to build, simulate, and validate PBPK models and perform virtual trials. |
Technical Support Center
This support center provides guidance for common issues encountered when integrating Quantitative Systems Pharmacology (QSP) and Machine Learning (ML) to refine Physiologically-Based Pharmacokinetic (PBPK) models, specifically within the context of predict-first research on hepatic first-pass metabolism.
Troubleshooting Guides & FAQs
Q1: During virtual population generation for a first-pass metabolism PBPK model, my ML-sampled parameters (e.g., CYP3A4 abundance, hepatic blood flow) lead to physiologically implausible combinations (e.g., high blood flow with low enzyme mass). How can I enforce biological constraints? A: This is a common issue with naive random sampling. Implement a conditional sampling or rejection sampling framework guided by known physiological correlations.
- Protocol: 1) Define joint distributions (e.g., from population databases). 2) Train a Generative Adversarial Network (GAN) or Variational Autoencoder (VAE) on real physiological data to learn latent relationships. 3) Generate virtual subjects from the latent space. 4) Include a rejection step: if sampled values fall outside predefined biologically feasible ranges (see Table 1), discard and resample.
- Key Check: Ensure your training data for the ML sampler encompasses the full covariance structure of human physiology.
Q2: When using ML to optimize QSP model parameters for first-pass prediction, the algorithm converges on a local minimum and fails to improve agreement with in vitro intrinsic clearance data. What steps should I take? A: This suggests issues with the optimization landscape or feature representation.
- Protocol: 1) Feature Engineering: Instead of raw parameters, use log-transformed or scaled parameters. Incorporate prior knowledge as Bayesian priors in the loss function. 2) Algorithm Switching: Combine global (e.g., particle swarm, genetic algorithm) and local (e.g., gradient-based) optimizers. Use the global method to explore broadly, then refine with a local method. 3) Ensemble Approach: Run multiple optimizations from different starting points. Analyze the cluster of best-fit parameter sets for identifiability issues.
Q3: My QSP-ML hybrid model predicts first-pass metabolism well for one drug class (e.g., CYP2D6 substrates) but generalizes poorly to another (e.g., UGT substrates). How can I improve model transferability? A: The model is likely overfitting to specific pathways. Implement transfer learning and modular QSP design.
- Protocol: 1) Retain the core physiological structure (blood flows, organ volumes) of the PBPK model. 2) For the new pathway, freeze shared ML layers and re-train only the task-specific layers (e.g., those predicting UGT1A1-mediated clearance) using a smaller dataset. 3) Use a multi-task learning setup during initial training where the ML component predicts multiple clearance pathways simultaneously, forcing it to learn more robust, generalizable representations of enzyme kinetics.
Q4: The final integrated QSP/ML model is a "black box." How can I extract explainable, mechanistic insights about first-pass metabolism for my research thesis? A: Employ Explainable AI (XAI) techniques specifically designed for hybrid models.
- Protocol: 1) Apply SHAP (SHapley Additive exPlanations) values to quantify the contribution of each input feature (e.g., lipophilicity, fu, isoform abundance) to the predicted first-pass extraction. 2) Use sensitivity analysis on the QSP model component by perturbing ML-predicted parameters and observing the change in output. 3) Perform partial dependence plots to visualize the relationship between a key ML-driven parameter (e.g., predicted transporter rate) and the overall first-pass metabolism.
Data Summary
Table 1: Key Physiological Parameters and Ranges for Virtual Population Generation in Hepatic First-Pass Models
| Parameter | Symbol | Typical Range (Healthy Adult) | Primary Source of Variability | Notes for ML Sampling |
|---|---|---|---|---|
| Hepatic Blood Flow | QH | 90 ± 15 L/hr | Body size, cardiac output | Correlate with cardiac output. |
| CYP3A4 Abundance | - | 20-150 pmol/mg microsomal protein | Genetics, age, induction/inhibition | Log-normal distribution. Correlate weakly with CYP2C9. |
| Portal Vein Blood Flow | QPV | ~75% of QH | Splanchnic hemodynamics | QH = QPV + Hepatic Artery Flow. |
| Microsomal Protein per Gram Liver | MPPGL | 30-50 mg/g | Disease state, donor variability | Critical for scaling in vitro clearance. |
| Hematocrit | HCT | 0.40-0.50 | Sex, health status | Impacts blood-to-plasma partitioning. |
Experimental Protocol: ML-Augmented In Vitro to In Vivo Extrapolation (IVIVE) for First-Pass Prediction
Objective: To refine the prediction of hepatic first-pass extraction (FH) by using ML to correct the systematic bias in traditional IVIVE.
Materials & Reagents:
- Substrate: Drug candidate(s) of interest.
- In Vitro System: Human liver microsomes (HLM) or hepatocytes (suspended/plated).
- Analytical: LC-MS/MS system for quantifying substrate depletion.
- Software: PBPK platform (e.g., GastroPlus, Simcyp, PK-Sim), Python/R with ML libraries (scikit-learn, TensorFlow/PyTorch).
Procedure:
- Generate In Vitro Clearance Data: Measure intrinsic clearance (CLint, in vitro) for a diverse set of ~20 known drugs with varying first-pass extraction.
- Obtain In Vivo Reference Data: Compile human in vivo clearance (CLH) and bioavailability (F) data for the same drugs from literature.
- Calculate Observed vs. Predicted Discrepancy: For each drug, use traditional QSP/PBPK scaling to predict FH from CLint, in vitro. Compute the residual error (ε = log(Predicted FH) - log(Observed FH)).
- Train ML Correction Model: Use molecular descriptors (logP, pKa, PSA), in vitro assay conditions, and predicted pathway involvement as features (X). The target variable (y) is the residual error (ε). Train a Random Forest or Gradient Boosting model.
- Apply to New Candidate: For a new drug, calculate its traditional IVIVE prediction, then use the trained ML model to predict the expected correction (εpred). Apply this correction: log(FH, corrected) = log(FH, traditional) - εpred.
- Validate: Use leave-one-out or external test set validation to assess improvement in prediction accuracy (e.g., RMSE, fraction within 2-fold error).
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Tools for QSP/ML-Enhanced First-Pass Metabolism Research
| Item | Function in Research |
|---|---|
| Human Hepatocytes (Cryopreserved) | Gold-standard in vitro system for measuring intrinsic clearance and transporter effects; provides full complement of metabolizing enzymes. |
| Recombinant CYP/UGT Enzymes | Used to deconvolute the contribution of specific isoforms to overall metabolic clearance. |
| PBPK/QSPSimulation Software | Platform for building the mechanistic physiological model and integrating ML-derived parameters or sub-models. |
| Python/R with ML Libraries | Environment for developing custom ML models for parameter prediction, virtual population generation, and bias correction. |
| Curated Clinical PK Database | Essential source of in vivo data for training and validating ML models (e.g., Pharmapendium, DrugBank). |
| SHAP/XAI Python Library | Critical for interpreting ML model outputs and deriving mechanistic hypotheses from black-box predictions. |
Visualizations
Title: ML-Augmented IVIVE Workflow for First-Pass Prediction
Title: QSP-ML Hybrid Model Architecture for First-Pass Metabolism
Technical Support Center
FAQs & Troubleshooting Guides for PBPK Modeling of First-Pass Metabolism
Q1: My PBPK model consistently underpredicts the oral bioavailability of a high-clearance drug metabolized by CYP3A4. What are the primary factors to investigate? A: This is a common issue. Systematically check the following, in order of likelihood:
- Gut Wall Metabolism: Ensure your model includes a compartment for the enterocyte (e.g., a "gut wall" compartment with defined volume and blood flow). Verify the scaling of CYP3A4 abundance (pmol/mg protein) from your in vitro data to the whole intestine, considering potential regional expression gradients.
- Hepatic Inflow Concentration: Confirm the model correctly calculates the concentration of drug reaching the liver, accounting for absorption rate and any concurrent gut metabolism.
- In Vitro to In Vivo Extrapolation (IVIVE): Review the scaling factors used. Key parameters include:
- Microsomal or hepatocyte binding (
fumicorfuhep) - Hepatic microsomal protein per gram of liver (
MPPGL) - Liver mass or blood flow
- Consider inter-system extrapolation factors (ISEF) for specific CYPs.
- Microsomal or hepatocyte binding (
- Transporter Effects: For substrates of efflux transporters like P-gp, confirm the
KmandVmax/CLintfor efflux are correctly parameterized, as this can limit access to gut enzymes.
Q2: How should I handle variability in enzyme abundance and activity when predicting population first-pass metabolism? A: To move towards personalized predictions, variability must be incorporated probabilistically.
- Define Distributions: Source population distributions for critical physiological parameters (see Table 1).
- Monte Carlo Simulation: Use these distributions to run virtual population trials (e.g., 1000 virtual subjects).
- Covariate Relationships: Implement known relationships (e.g., CYP3A4 abundance vs. age, CYP2D6 activity vs. genotype).
Table 1: Key Variability Parameters for Population PBPK of First-Pass Metabolism
| Parameter | Typical Distribution (Example) | Source/Justification | Impact on First-Pass |
|---|---|---|---|
| CYP3A4 Abundance (Liver) | Log-normal (CV ~40%) | Proteomic data from tissue banks | Directly scales hepatic CLint |
| CYP3A4 Abundance (Duodenum) | Log-normal (CV >60%) | Higher variability than liver | Major driver of gut wall variability |
| Liver Volume | Normal (CV ~10%) | Population imaging studies | Affects organ clearance capacity |
| Hepatic Blood Flow | Normal (CV ~15-20%) | Physiological literature | Determines hepatic delivery rate |
| Enterocyte Blood Flow | Assigned as fraction of cardiac output | System model | Influences gut wall extraction |
| Plasma Protein Binding (fu) | Log-normal or Beta | Clinical data | Affects free concentration for metabolism |
Q3: What is a robust experimental protocol to generate in vitro data for gut metabolism parameters?
A: Protocol for Determining Intrinsic Clearance (CLint) in Human Intestinal Microsomes (HIM)
- Objective: To obtain
CLint, gutfor IVIVE to gut wall metabolism. - Materials: Test compound, pooled HIM (from specific intestinal regions), NADPH regeneration system, phosphate buffer (pH 7.4), methanol/acetonitrile (stop solvent).
- Procedure:
- Prepare incubation mixtures (n=3) containing HIM (0.2-0.5 mg protein/mL), test compound at a concentration <<
Km(typically 1 µM), and buffer. - Pre-incubate for 5 min at 37°C.
- Initiate reaction by adding NADPH system.
- Aliquot at 7-8 time points (e.g., 0, 3, 6, 9, 15, 20, 30, 45 min) and quench with cold organic solvent.
- Centrifuge, analyze supernatant via LC-MS/MS to determine parent compound depletion.
- Include controls without NADPH and without microsomes.
- Prepare incubation mixtures (n=3) containing HIM (0.2-0.5 mg protein/mL), test compound at a concentration <<
- Data Analysis: Plot Ln(% parent remaining) vs. time. The slope (
k) is the depletion rate constant. CalculateCLint, gut=k/ (mg protein per mL incubation). Scale to whole intestine using total mg microsomal protein in the gut.
Q4: The model fails when simulating a drug that is both a CYP3A4 substrate and a P-gp victim. How can I model this interaction? A: This requires a defined compartmental structure that allows for sequential/pericellular transport and enzyme interaction. Implement a "gut wall" compartment with simultaneous mass-action equations for influx, efflux (P-gp), and metabolism (CYP3A4). The diagram below illustrates the logical relationship and required parameters.
Diagram: Gut Wall Disposition Logic for Dual Substrates
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in First-Pass PBPK Research |
|---|---|
| Pooled Human Liver Microsomes (HLM) | Contains major CYPs for determining hepatic CLint. Essential for IVIVE of liver metabolism. |
| Pooled Human Intestinal Microsomes (HIM) | Contains intestinal CYP isoforms (esp. CYP3A4) and UGTs. Critical for quantifying gut wall metabolism. |
| Recombinant CYP Enzymes (rCYP) | Used for reaction phenotyping to identify which specific enzyme(s) metabolize the drug. |
| Transfected Cell Systems (e.g., Caco-2, MDCK with P-gp/BCRP) | Determine permeability (Papp) and active transporter kinetics (Km, Vmax) for gut absorption models. |
| Cryopreserved Human Hepatocytes | Gold standard for hepatic CLint, incorporating uptake, metabolism, and efflux in an integrated cellular system. |
| Physiologically Relevant Buffer Solutions | Simulate intestinal pH gradients (FaSSIF/FeSSIF) for accurate solubility and dissolution input parameters. |
Q5: What workflow should I follow to develop a predictive PBPK model for virtual bioequivalence (VBE) of a modified-release product? A: VBE requires a model that accurately captures dissolution, regional absorption, and regional first-pass extraction. Follow this integrated workflow.
Diagram: PBPK Workflow for Virtual Bioequivalence
Conclusion
PBPK modeling represents a transformative, mechanistic approach for predicting first-pass metabolism, offering significant advantages over empirical methods in the design and development of oral drugs. By grounding models in robust physiological and biochemical data, researchers can more accurately forecast oral bioavailability, identify potential formulation challenges, and reduce late-stage attrition. Future advancements lie in the integration of quantitative systems pharmacology (QSP), refined virtual population generators, and enhanced in vitro-in vivo extrapolation (IVIVE) techniques. As regulatory acceptance grows, the strategic application of validated PBPK models will continue to accelerate rational drug design, optimize clinical trial planning, and pave the way for more predictable and successful therapeutic outcomes.